为什么要标准化?
简单讲,就是为了让一个特定的突变具有一个唯一的表述方式,一个突变如果不进行标准化,会出现多种不同的表达方式,即MNP(Multi Nucleotide Polymorphism ),这会给大家的交流带来诸多的问题, 试想每个人有自己的习惯,最终一个突变可能会有成百上千种表达方式,这无疑会给研究人员交流带来诸多的困扰。因此标准化是很有必要的,目前主流的标准化原则是涉及尽可能少的碱基,尽可能做对齐。
首先我们来看一下,目前的变异检测多是依靠序列比对,然后会针对不同的比对情况进行变异检出的,我们先来看一下SNV, 如果不标准化,一个相同的变异 CAT => TGC 的突变,会出现下列所示的多种不同的表述方法。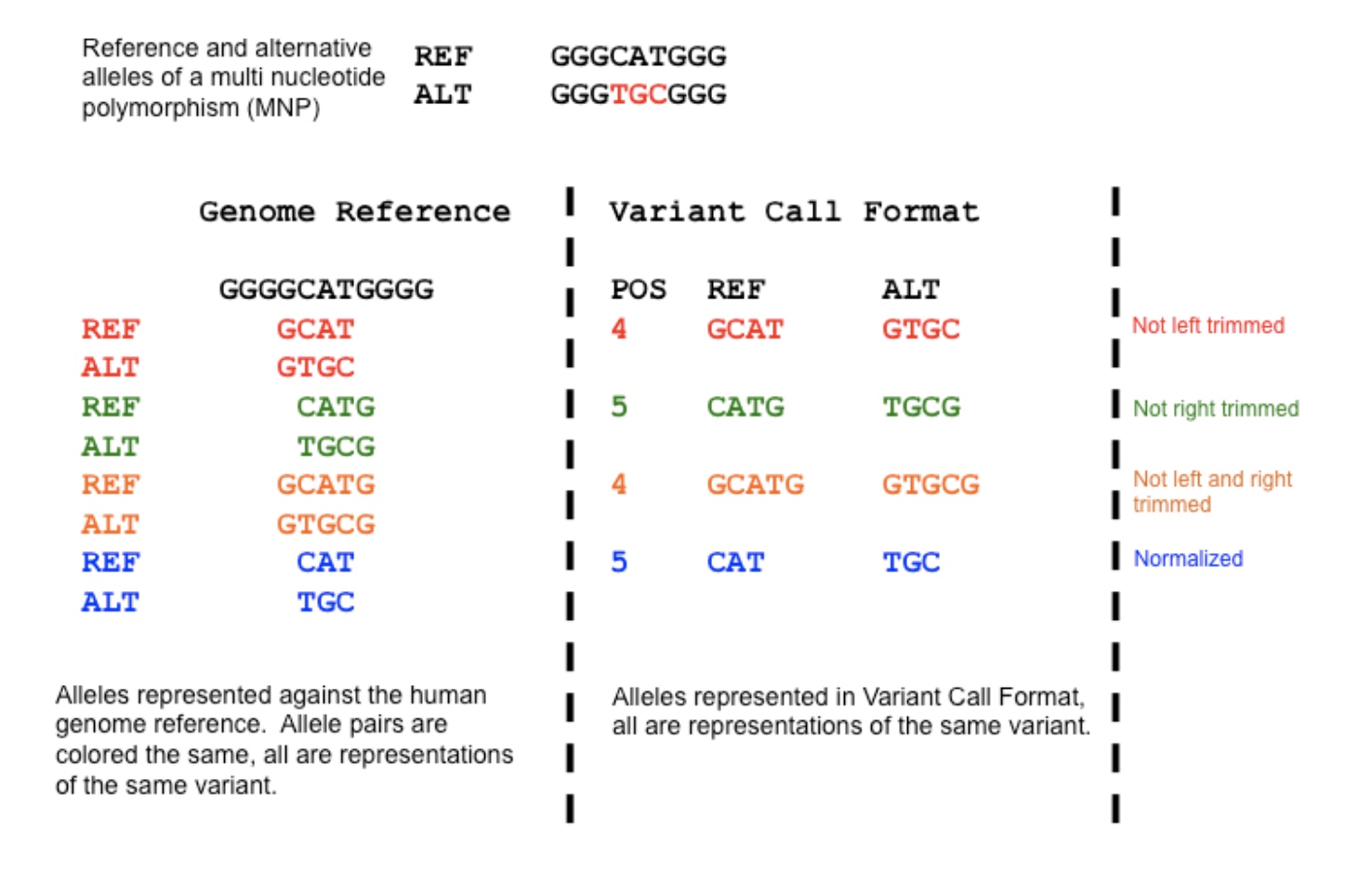
随着基因检测技术的广泛应用,越来越多与疾病相关的基因和变异被明确。为了让变异注释更程序化、对变异文献的检索更全面化以及对变异的描述更标准化故需要形成统一突变命名规则,HGVS命名规则是目前公认的命名规则(人类基因组变异学会(HGVS)、人类变异项目(HVP)和人类基因组组织(HUGO)授权)。
HGVS命名规则从不同层面对变异进行描述,反应变异的位置及对编码的蛋白的影响。参考序列的选择、转录版本号的不同及描述层面(DNA水平、RNA水平及蛋白水平)的不同,均会导致同一变异描述的形式不同。
HGVS规范
参考序列
只接受NCBI或EBI公共数据库中的参考序列ID,且必须同时包含accession和version信息;如NC_000023.10中,NC_000023代表accession号,10代表version号。参考序列中下划线前的大写字母代表参考序列格式,目前批准的参考序列格式有:
|格式|解释|举例|
|-|-|-|
|NC_#|代表完整的基因组序列,标记的类别包括基因组、染色体、细胞器、质粒。|NC_000023.10:g.32407761G>A|
|LRG_#|Locus Reference Genomic,基因座参考基因组序列。(不能在其他基因组找到的)|NG_012232.1:g.954966C>T|
|NG_#|不完整的基因组区域(不转录的假基因或者那些很难自行化注释的基因组区域)|LRG_199:g.954966C>T|
|NM_#|编码蛋白的转录本序列。基因检测报告中最常用此作为参考序列。|NM004006.2:c.4375C>T|
|NR_#|非编码的转录本序列,包括结构RNAs,假基因转子等。|NR002196.1:n.601G>T|
|NP_#|蛋白质序列|NP-003997.1:p.Arg1459*(p.Arg1459Tero|
- g.代表线性基因组参考序列;
- o.代表环状基因组参考序列;
- m.代表线粒体参考序列;
- c.代表编码DNA参考序列;
- n.代表非编码DNA参考序列;
- r.代表RNA参考序列;
- p.代表蛋白(氨基酸)参考序列。
变异位置
- 编码区(CDS)
- 以起始密码子ATG的第一个碱基A开始,并记为c.1,以终止密码子(TAA, TAG, TGA)的最后一个碱基为终点。
- 内含子区(Intron)
- 靠近内含子5’末端的变异位点,需依据上游最近外显子的最后一个碱基来定位,如c.87+4,代表上游最近外显子的边界位置为87,变异位点在内含子5’ 端开始的第4个碱基;
- 靠近内含子3’ 末端的变异位点,要依据下游最近外显子的第一个碱基来定位,如c.88-11,
- 内含子碱基个数为偶数时,中间碱基平分后按上下游外显子碱基来定位命名,如…,c.87+676, c.87+677, c.87+678, c.88-678, c.88-677, c.88-676, …
- 内含子碱基个数为奇数时,中间碱基相对于上游外显子最后一个碱基来定位命名,如…,c.87+677, c.87+678, c.87+679, c.88-678, c.88-677, …
- 非编码区(UTR区):
- 起始密码子ATG上游(5’ UTR区)标记为“-”,编号为c.-1, c.-2, c.-3…
- 终止密码子下游(3’ UTR区)标记为“”,编号为c.1, c.2, c.3…
- 位于靠近5’ UTR和3’ UTR区的内含子变异位点,命名规则同内含子区,如:5’ UTR区内含子为c.-85+1,c.-84-3等;3’ UTR区内含子为c.37+1,c.38-3等。
参考示意图如下: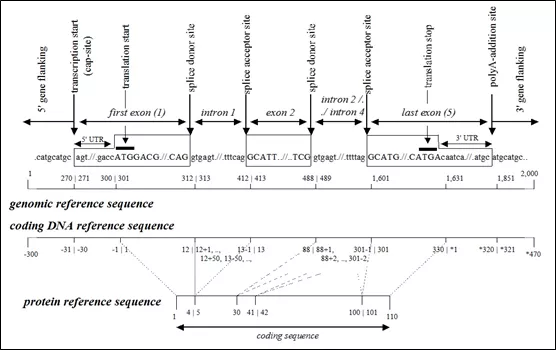
变异类型
优先级从高到低分别如下:
- 置换(>):一个核苷酸被另一个核苷酸替代,使用“>”来表示;例如g.1318G>T;
- 缺失(del):一个或多个核苷酸被移除,使用“del”进行描述;例如g.3661_3706del;
- 倒置(inv): 与原始序列反向互补的新的核苷酸序列(大于1个核苷酸)替换原始序列,例如由CTCGA变为TCGAG,使用”inv“表示;
- 重复(dup):一个或多个核苷酸拷贝直接插入原始序列的下游,使用“dup”表示;
- 插入(ins):序列中插入一个或多个核苷酸,并且插入序列并非上游序列拷贝;
- 缺失-插入(delins/indel):一个或多个核苷酸被其他核苷酸替代,但并不是发生替代、倒置和转置;
- 转换(con):一种特殊类型的缺失-插入,其中替代原始序列的核苷酸序列是来自基因组中另一个位点的序列拷贝;
5、变异描述示例
DNA水平
- c.76A>C:76位的核苷酸A变异为C;
- c.82_83delTG:位于82和83位点上的核苷酸TG缺失,ACTTTGTGCC变异为ACTTTGCC(A是第76位);
- c.83_84dupTG: ACTTTGTGCC(A为第76位)的83-84位之间插入短的串联重复序列TG,变为ACTTTGTGTGCC;
- g.333_590con1844_2011:基因组中编号为333-590的核苷酸序列替代1844-2011原有序列,插入其中;
- g.112_117delinsTG:在基因组序列编号为112-117之间的6个核苷酸被TG替换;
多个变异使用”[]”标注变异,并用“;”链接 - 同一等位基因发生多个变异:
c.[76A >C;83G>C]:同一染色体上76位和83位发生两个变异(顺式); - 不同等位基因发生多个变异:
c.[76A >C];[83G>C]:两个变异发生在不同染色体上(反式); - 不确定多个变异发生的位置:
c.[76A >C](;)[83G>C]:两个变异可能发生在同一染色体,也可能发生在不同染色体上,用(;)来链接; - 定义重复序列的核苷酸范围及重复单位的数量,并用“[]”表示
g.123_124[4]:基因组序列中第123-124间的核苷酸重复出现4次; - 对于短的/简单的重复,可以展示重复序列
g.123TG[4]:基因组序列中从123位开始TG核苷酸重复出现4次; - 当重复序列长度不确定时,使用括号进行指定
g.-128GGC[(600-800)]:基因组编码区上游128位核苷酸处重复插入GGC,重复次数在600-800之间;蛋白质水平
- 替换:如p.Trp26Cys,表示第26位的Trp被Cys取代(错义突变);p.Trp26Ter (p.Trp26*),表示第26位的Trp变为终止密码(无义突变);p.Cys123=,表示基因突变之后,氨基酸没有发生改变(同义突变);
- 缺失:如p.Ala3_Ser5del,表示多肽序列中从第3位的Ala到第5位的Ser发生了缺失;
- 插入:如p.Lys2_Gly3insGlnSerLys,表示在第2位的Lys和第3位的Gly之间插入了GlnSerLys;
- 插入缺失:如p.Cys28delinsTrpVal,表示第28位的Cys缺失,同时被TrpVal取代;
- 重复:如p.Ala2[10],表示第2位的Ala重复了10次;
- 移码突变:在起始密码子和终止密码子之间的读码框发生了改变;以“fx”进行表示;如p.Arg97ProfsTer23,表示第97位的Arg是首个发生改变的氨基酸,且Arg变为Pro,同时发生移码突变后,终止密码的位置变为第23位;
变异类型的描述原则
一般原则:
- 所有变异需先从最基本的层面(DNA水平)进行描述,还可从RNA水平和蛋白质水平上进行描述。
- 用变异的描述是否加“()” 来说明变异是由实验确定的还是从理论上推导出来的。
- 所有的变异都应该根据公认的参考序列来描述。
- 当变异可描述为几种变异类型时,优先级为:(1)替换,(2)删除,(3)倒位,(4)重复,(5)转换,(6)插入。
如:当一个变异可以被描述为重复或插入时,根据优先级决定应描述为重复,而不是插入。
如:“-ATGCCCA-”插入后变为“-ATGCCCCA-”,应描述为c.7dup,而不是NM_004006.2:c.7_8insC。 - 在进行变异描述时,基因的描述要采用HGNC的官方基因名。
重复序列变异描述原则:c.123CAG[16],g.3258796GA[8]
对于编码区DNA序列而言,重复序列的描述仅用于重复单元长度为3的倍数的重复序列,即不会影响阅读框的重复单元长度;若重复序列长度不是3的倍数,则不能用该形式描述。
如:当CNPTAB基因在编码区的TATA序列后插入TATATATA序列,则对于该插入变异的描述应为NM_024312.4:c.1741_1742insTATATATA,而不是NM_024312.4:c.1738TA[6]。
3’ 端法则:变异的描述需遵循最靠近3’ 端法则。
如“-ATGCCCCA-”变异成“-ATGC_CCA-”,根据3’ 端法则应描述为c.7delC,而不是c.5delC。
例外:当缺失/重复发生在外显子与外显子衔接处,且衔接处碱基相同,不遵循3’ 端法则。
如“..GAT gta..//..cag TCA..”缺失后变为“..GA_ gta..//..cag TCA..”,应描述为NM_004006.2:c.3921del,而不是NM_004006.2:c.3922del;“..GAT gta..//..cag TCA..”重复后变为“..GATT gta..//..cag TCA..”,应描述为NM_004006.2:c.3921dup,而不是NM_004006.2:c.3922dup。
- 有大量的原始检测软件,在未进行变异注释前是进行的左对齐

delins原则:涉及两个或以上连续核苷酸的替换,描述为delins。
若两个变异被一个或多个核苷酸分隔,优先单独描述两个变异,而不采用delins合并描述;若被一个核苷酸分隔的两个变异,共同影响一个氨基酸,则合并描述为delins,如c.142_144delinsTGG (p.Arg48Trp);
若两个变异中的任何一个为已知的高频变异位点,则需要单独描述两个变异,即NM_004006.1:c.[145C>T;147C>G],优先于NM_004006.1:c.145_147delinsTGG。(该原则需与解读同事讨论)
起始密码子变异描述:描述取决于变异对蛋白产物改变的结果。
- 变异后不产生蛋白质:NM_003002.3:p.0
- 变异对蛋白产物的影响不清楚且无法预测:NM_003002.3:p.?
- 变异后产生新的起始氨基酸:
a)上游:p.Met1ext-5,即原始密码子上游第5位(5’ UTR区)产生了新的起始氨基酸,另可描述为p.Met1extMet-5。
b)下游:p.Leu2_Met124del,即原起始氨基酸丢失且下游产生新的起始氨基酸,导致蛋白的第1到123位氨基酸缺失。终止密码子变异描述:采用“ext”描述
- p.Ter110Glnext17(p.110Glnext17):原终止氨基酸变为谷氨酰胺(Gln),并在下游第17位产生新的终止氨基酸,导致蛋白产物延长17个氨基酸。
注:不可描述为p.Ter110GlnextTer17 ,此处的17代表的是位置(3’ UTR区) - p.Ter315TyrextAsnLysGlyThrTer(p.315TyrextAsnLysGlyThr) :原终止氨基酸变为酪氨酸(Tyr),并在下游第5位产生新的终止氨基酸,导致蛋白产物延长5个氨基酸。
- p.Ter327Argext?(p.327Argext*?):原终止氨基酸变为精氨酸(Arg),导致蛋白产物延长,延长的长度未知。
附录
1、特殊字符的含义
- “+”:c.123+45A>G(代表靠近内含子5’ 端的核苷酸发生变异)
- “-”:c.124-56C>T(代表靠近内含子3’ 端核苷酸发生变异);c.-142C>G(代表5’ UTR区)
- “”:c.32G>A(代表3’ UTR区);P.Trp41*(代表终止氨基酸)
- “[]”:代表等位基因,“;”用来分隔变异和等位基因,如g.[123456A>G;345678G>C] 代表顺式,g.[123456A>G];[345678G>C]代表反式, g.123456A>G(;)345678G>C代表这两个变异的顺式反式未知。
- “()”:用来表示不确定的或预测的结果,如p.(Ser123Arg)
- “?”:用来表示核苷酸或氨基酸的位置未知,如g. (?_234567) _ (345678_?)del
- “^”:代表“或”的意思,如p.Ser124Arg反推核苷酸的改变为c.(370A>C^372C>G^372C>A) ,即AGC变成CGC, AGG或AGA
- “/”:表示嵌合体(mosaic),如NM_004006.1:c.145=/C>T
- “//”:表示异源嵌合体(chimeric),如NM_004006.1:c.85=//T>C
- “|”:代表不是序列的直接改变,而是一种修饰或一种状态的改变,如甲基化。
- “::”:用于描述RNA融合转录本和断点连接形成的环状染色体
缩写字符的含义
- “fs”代表变异类型为移码变异,主要是针对蛋白水平而言,如p.Arg456GlyfsTer17或p.Arg456Glyfs*17
- “ext”代表变异类型为延长,主要是针对起始密码子和终止密码子变异导致的蛋白水平的改变,如p.Met1ext-5
- “gom” 表示获得甲基化,如g.12345678_12345901|gom
- “lom”表示去甲基化,如g.12345678_12345901|lom
- “met” 表示甲基化,如g.12345678_12345901|met=
软件脚本
3’对齐
针对变异进行3’对其的工具,可以参考GitHub仓库的脚本 toolkits\03.Deal_mutation\Realn4vcf.py
左对齐
目前仍有大量软件本身是进行左对齐的,因此在进行结果比较时可以统一进行左对齐,从而实现变异结果的规范化,进行比较。
对齐的命令行1
2
3
4
5
6
7
8
9
10
11Commands used are:
bcftools norm -f ref.fa in.vcf -O z > out.vcf.gz
java -jar GenomeAnalysisTK.jar -T LeftAlignAndTrimVariants --trimAlleles -R ref.fa --variant in.vcf.gz -o out.vcf.gz
vt normalize -r ref.vcf.gz -o out.vcf.gz
Versions are:
bcftools v0.2.0-rc8-5-g0e06231 (using htslib 0.2.0-rc8-6-gd49dfa6) [updated non release development version]
GATK v3.1-1-g07a4bf8
vt normalize v0.5
参考资料: WIKI
HGVS
HGVS Recommendations for the Description of Sequence Variants: 2016 Update