参考文献
Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer
针对上述指南,在2017年,发布翻译文章发表在中国科学杂志 遗传变异分类标准与指南
整理汇总PPT材料
摘要
下一代基于测序的癌症测试的临床实验室的广泛实施突出了标准化实验室间分子结果的解释和报告的重要性和潜在的好处。分子病理学协会召集了一个多学科工作组,负责评估基于下一代测序技术的癌症检测的现状,并建立体细胞序列变异的标准化共识分类,注释,解释和报告惯例,并由美国医学遗传学和基因组学学院联络代表,美国临床肿瘤学会和美国病理学家学院。根据专业调查,文献综述和工作组主题专家共识的结果,提出了一种基于体细胞序列变异的临床意义进行分类的四级系统:一级,具有很强临床意义的变异; II级,具有潜在临床意义的变体; III级,临床意义未知的变体;和第四级,变体被视为良性或可能良性。癌症基因组学是一个快速发展的领域。因此,应不断评估治疗,诊断或预后中任何变异的临床意义。报告基因组变异应遵循标准命名法,并明确描述测试方法和限制。临床建议应简明扼要,并与组织学和临床发现相关联。
分类
- tier I, variants with strong clinical significance;
- tier II, variants with potential clinical significance;
- tier III, variants of unknown clinical significance;
- tier IV, variants deemed benign or likely benign.
导读
基于二代测序技术的肿瘤检测已越来越多的应用于临床实验室中,但目前在不同实验室间存在检测方法、报告内容等方面的差异,这对遗传检测的解读以及普及应用造成了一定的影响。因此,在不同实验室间建立统一的分子检测结果的解读和报告标准,及建立行业标准,显得尤为迫切。
在2015年春天,在美国专门成立了一个以临床实验室为核心的工作组,其组成包含了分子病理协会(AMP)、美国医学遗传学与基因组学协会(ACMGG)、美国临床肿瘤学会(ASCO)与美国病理学家协会(CAP)的一线专家,该工作组的主要工作为对肿瘤及疑似肿瘤相关的序列变异检测建立检测标准并在行业达成共识。
该工作组首先对北美地区超过40家的临床检测实验室进行了问卷调查,结果显示不同实验室在检测组织类型、检测基因数量、是否检测肿瘤组织全外显子组或全基因组、以及其他细节方面都存在较大差别,此外在不同单位的检测报告的报告内容方面也存在较多差异。该工作组认为,为医疗机构提供分类的遗传变异报告对病人及整个医疗行业都极为重要,包括:提供精确的肿瘤对靶向治疗反应性信息;建立国家级别的医疗指南;以及与临床试验合作,对建立不同实验室间的通用标准提供支持。基于以上这些考量,工作组专家们根据已有数据、文献报道和专业知识,给出以下指南建议.
对多个公司的报告情况调研结果如下
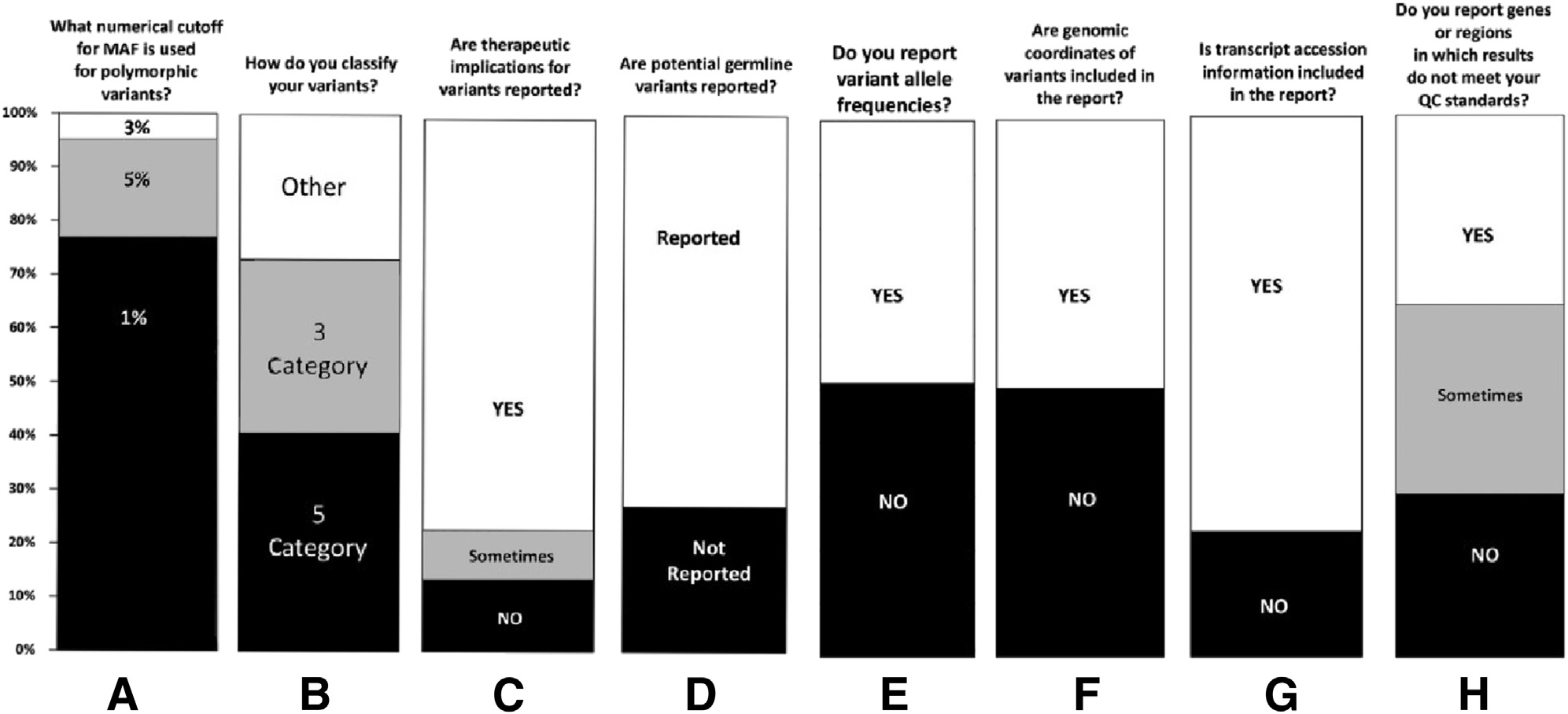
图1 AMP对NGS技术及NGS结果解读的调研 A: MAF阈值. B: 变异分类数目 C:报告中是否包含治疗性建议 . D: 报告中是否包含潜在的生殖细胞突变. E: 报告是否包含变异等位频率Variant allele frequency (VAF) F: 报告是否包含基因组坐标 G: 报告是否包含转录本ID(Transcript accession) H: 报告是否包含不符合质控的基因/区间
数据库
Genomic Databases
随着越来越多的针对各种肿瘤类型的大规模基因组测序项目的发布,全球正在产生大量的基因组信息并将其整合到许多公共数据库中(表1)。例如,美国国家癌症研究所(National Cancer Institute)的基因组数据共享(Genome Data Commons)包含美国国家癌症研究所(National Institutes Institutes)从一些最大和最全面的癌症基因组数据集中生成的数据.包括 :
The Cancer Genome Atlas, Therapeutically Applicable Research to Generate Effective Therapies, and the Cancer Genome Characterization Initiative (https://gdc.cancer.gov, last accessed September 25, 2016). Another public somatic variant database is the Catalog of Somatic Mutations in Cancer (http://cancer.sanger.ac.uk/cosmic, last accessed September 30, 2016).
其中包含数百万种跨多种肿瘤类型的体细胞变异。体细胞变异分析中经常使用的其他几个数据库,例如参考序列信息,种群数据库和种系变异数据库也在不断增加和改进。基因组数据库提供了准确注释和确定体变异优先级所必需的信息。
在使用这些数据库是,整体上应该遵循乳癌规则:
- 了解数据库的内容以及如何汇总数据。临床实验室应查看与给定数据库有关的文档或公开文献,以确定数据库的来源,类型和意图
- 特别注意每个数据库的限制,以避免对注释结果的过度解释。
- 确认人类基因组装配的版本以及mRNA转录本参考,以确保适当的人类基因组变异学会(HGVS)注释。
- 尽可能使用基因组坐标而不是HGVS命名法来明确查询基因组数据库。
- 根据出版物或其他数据库的来源,单个或多个特定条目的数量,研究的深度,使用适当的对照,确认变异的体细胞来源,评估提供的基因组数据的质量以及功能和潜在药物反应研究。
- 验证所提供病理诊断的数据质量(例如,地点,诊断和子类型
Reference Sequence Databases
参考序列数据库提供有关人类基因组装配版本的信息以及相关信息,例如基因组坐标,以明确表示序列变异。附加信息,例如mRNA转录本的登录和版本(例如,BRAF NM_004333.4)和外显子边界定义,对于产生变异的正确HGVS命名法至关重要。可以从这些数据库中计算基因的变异位置图谱(编码,非编码,非翻译区和剪接位点)和链表述(阳性与阴性)。这也允许在没有基因组坐标信息的情况下明确表示变异。一些常用资源包括:
- RefSeq(国家生物技术信息中心参考序列数据库,https://www.ncbi.nlm.nih.gov/refseq,上次访问时间为2016年1月2日)
- Ensembl(http://www.Ensembl.org)。 ensembl.org/index.html,最后访问时间为2016年1月2日)
- Locus Reference Genomic (https://www.lrg-sequence.org,最近访问时间为2016年2月2日)。
Population Databases
这些数据库提供了有关大量特定人群中给定基因座上替代(次要)等位基因频率的全面信息。这些数据库通常用于根据次要等位基因频率(MAF)的任意临界值筛选出被认为是多态/良性的变异。目前尚无用于去除多态或良性变异的MAF的标准临界值。在没有正常组织配对的情况下,工作组建议使用1%(0.01)作为主要阈值,这在许多临床实验室中也很普遍。尽管总体MAF最常用,但临床实验室可能会考虑使用种族-根据患者的种族背景确定特定的MAF。在解释体细胞变异时,必须谨慎使用这些数据库,因为在参与研究时,假定参与这些测序研究的个体是健康的或没有亚临床疾病。确实,一些众所周知的经典癌症相关的和可靶向的体细胞改变已作为种群数据库的种系变异包括在内。例如,变异NM_004972.3(JAK2):c.1849G> T
(c.V617F)通常被看作是叶绿体增生的体细胞变异体
肿瘤,可以用FDA批准的Janus激酶(JAK)抑制剂靶向。它也包含在多个人群数据库中,例如 The Database of Short Genetic Variation (the National Center for Biotechnology Information database of genetic variation), Exome Variant Server, and Exome Aggregation Consortium(表格1)。在评估可能的血液系统恶性肿瘤时应格外小心,因为白血病和骨髓增生异常综合症中的许多常见突变基因也可能在其他健康个体的血液中发生体细胞突变,因此可能被错误地注释为多态性。
Cancer-Specific Databases
这些数据库提供了有关不同癌症和亚型谱中序列变异的发生率和普遍性的信息,对其他基因组数据库的交叉引用以及对已发表或未进行系统综述的文献的引用,细胞途径,靶向疗法,临床试验 以及结果数据。 从这些数据库中提取的不同癌症中的序列变异体的普遍性和分布,应谨慎解释,因为病理诊断标准的代表性较差,缺乏临床级别的文献管理以及提交变异体的来源控制不严(例如,探索性或 发现研究)。 例如,这些数据库中包括一些常见的种系良性变异,例如 the Catalog of Somatic Mutations in Cancer database 中的NM_000222.2(KIT):c.1621A> C(p.M541L)。 表1列出了常用的体细胞变异数据库。
Constitutional Variant Databases
常见的是,在有或没有匹配的正常组织的情况下进行肿瘤测序可能会揭示出种系起源的变异,例如与癌症易感综合症相关的基因中的致病变异。种系突变数据库,例如人类基因突变数据库和其他疾病或基因座特异性突变数据库,是评估这些变异的有用资源。这些数据库也可用于评估在这些数据库中报道了经过充分研究的种系对应物的体细胞变异(例如,TP53和PTEN基因中的某些变异)。另一个常用的数据库是ClinVar(http://www.ncbi.nlm.nih.gov/clinvar)。 ClinVar处理所有种类的稀有种系变异,例如病原体和良性,并在可用时提供相关的临床和实验证据。专家小组对ClinVar中的某些变异进行了有关其致病性的审查。目前,该数据库仅宿主种系变异,并有望在不久的将来纳入体细胞变异。
Internal (Laboratory-Generated) Databases
需要强调的是,临床实验室应该建立一个标注良好的内部数据库,以跟踪实验室中识别出的变异并提供一致的变异注释。这样的数据库可用于识别可能由测序比对伪像引起的潜在假阳性呼叫,以及确定实验室通常遇到的癌症类型的突变频率。我们强烈鼓励体细胞变异数据共享,并敦促临床实验室将精心挑选的变异体贡献到公共变异数据库中,以促进对体细胞变异体的准确解释。但是,此类提交过程应标准化并符合联邦隐私法规,即《健康保险可移植性和责任法案》以及《经济和临床健康信息技术法案》(原文 the Health Insurance Portability and Accountability Act and
the Health Information Technology for Economic and Clinical
Health Act.)。 正在努力建立临床级基因组数据库
In Silico (Computational) Prediction Algorithms

在计算机模拟中,预测算法是预测基因中核苷酸变化是否会改变蛋白质结构和功能的常用工具(表2)。起初,开发了早期常用算法并验证了用于胚系变异的算法。随后,外推它们的用途以解释体细胞变异。尽管各个算法的核心风险预测方法可能有所不同,但是它们可以分为两类:错义变异对蛋白质功能的影响的预测和序列变异对剪接的影响。考虑到氨基酸或核苷酸残基的进化保守程度,特定理化特性的氨基酸取代的生物化学影响以及变异在翻译蛋白质中的位置,是不同算法用来预测功能的一些主要标准错义变异的影响。拼接位点预测算法使用各种统计方法,例如马尔可夫模型,机器学习(神经网络)和最大熵原理,来预测变异是否会对拼接产生任何影响。通常,错义和剪接位点预测工具具有中等的特异性(大约60%至80%),并且倾向于过度预测有害影响。
在癌症基因功能的背景下,对这些预测的解释通常并不直接了当,特别是对于激活突变。例如,当通过多种计算机模拟算法进行分析时,经典的BRAF V600E致癌突变结果会具有冲突甚至良性的作用。这是临床实验室在进行体细胞变异解释时应该意识到的几种情况之一,同时在解释计算机分析的评分结果时应谨慎行事。建议不要将这些预测算法的结果用作变异分类或临床决策的唯一依据。
Variant Identification and Annotation
变异检测是变异解释的关键起点。 有许多变异检测软件工具(Supplemental Table S2).可以满足一种特定的检测,例如SNV,插入缺失,结构变异和CNV
(Supplemental Table S1):
| Variant caller | Location (URL) |
|---|---|
| MuTect v1.1.555 | https://www.broadinstitute.org/cancer/cga/mutect |
| Genome Analysis Toolkit (GATK) – MuTect v2s | https://www.broadinstitute.org/gatk/guide/tooldocs/org_broadinstitute_gatk_tools_walkers_cancer_m2_MuTect2.php |
| VarScan 256 | http://dkoboldt.github.io/varscan/ |
| VarDict57 | https://github.com/AstraZeneca-NGS/VarDict |
| Sterlka58 | https://sites.google.com/site/strelkasomaticvariantcaller/ |
| FreeBayes59 | https://github.com/ekg/freebayes |
| Scalpel60 | http://scalpel.sourceforge.net/ |
| Pindel61 | http://gmt.genome.wustl.edu/packages/pindel/ |
| SAMtools62 | http://samtools.sourceforge.net/ |
| Torrent Suite Variant Caller | https://github.com/iontorrent/TS |
| SomaticSniper63 | http://gmt.genome.wustl.edu/packages/somatic-sniper/ |
对于临床实验室来说,了解这些变异检测工具的局限性很重要。 对生物信息学流程(包括商业购买的生物信息学软件包)进行适当的实验室验证对于确保结果的质量至关重要。 某些称为变异的指标对于变异解释至关重要,例如 supporting reads (depth of coverage) and variant allele frequency (VAF),应纳入变异体评估中; 后者对于在没有配对正常的情况下的体细胞变异解释和评估肿瘤克隆多样性特别重要。
变异检测结果一般使用一些标准格式进行输出展示,例如clinical variant call format(VCF), genomic VCF 和 general feature format (alias gene-finding format or generic feature format).
The VCF is the most widely used schema in the clinical laboratories as of 2016 to represent detected variants (Clinical Variant Call Format, http://vcfclin.org, last accessed September 28, 2016). Required VCF fields include genomic coordinates, reference nucleotide(s), and variant nucleotide( s). However, complex, multinucleotide, and large structural variants are difficult to represent in the current specification of VCF file format version 4.2, despite the ongoing efforts for standardizing variant representation.
For further clinical interpretation, additional metadata that add meaningful and readily identifiable information to variants should be included (eg, gene symbol, variant location, variant type, HGVS nomenclature for cDNA sequence changes, and predicted protein sequence alterations). Additional resources, such as cross-references to external databases (cancerspecific and general genomic databases) (Table 1) and precomputed in silico algorithm-based predictions (Table 2), can also be beneficial. This process is formally referred to as variant annotation, and may be automated by software tools(Supplemental Table S2).
(Supplemental Table S2):
| Software | Location (URL) |
|---|---|
| Annovar64 | http://annovar.openbioinformatics.org/en/latest/ |
| snpEff65 | http://snpeff.sourceforge.net/ |
| SeattleSeq | http://snp.gs.washington.edu/SeattleSeqAnnotation138/ |
| AnnTools66 | http://anntools.sourceforge.net/ |
| NGS-SNP67 | https://www.ualberta.ca/~stothard/downloads/NGS-SNP/ |
| VEP (Variant Effect Predictor)15 | http://useast.ensembl.org/info/docs/tools/vep/index.html |
变异注释对于准确解释体细胞序列变异至关重要。这些对变异注释得到的多元数据构成了变异评估和解析的初始内容。
变异注释面对的一个重要挑战就是将基因组坐标(即染色体和位置)转换为相应的cDNA /氨基酸坐标系统(分别为c.和p.syntax)以进行解读。
这个问题针对indel变异由于对齐排列导致变异表述不一致的问题上,表现尤其突出。尽管HGVS系统建议使用右对齐表示法(将变异的开始位置向右移动,直到不再可能这样做),但VCF规范要求使用左对齐表示法。当前可用的注释解决方案仅部分解决了该问题。缺乏左/右对齐的标准化可能会严重影响变异定位,从而导致变异命名错误。根据HGVS命名法,当存在多个替代转录本时,必须使用正确的mRNA转录本编号和版本信息,以确保变异描述的准确和一致。临床实验室在内部数据库中使用变异的基因组位置来存储变异信息也非常重要,以确保数据存储的明确和可回查。
Proposed Guideline for Evidence-Based Categorization of Somatic Variants
体细胞变异包括SNV,插入缺失,基因组重排产生的融合基因和CNV。与胚系变异的解释(侧重于特定疾病的变异性或疾病因果关系)不同,体细胞变异的解释应着重于其对临床的影响。如果变异预测对特定疗法的敏感性,耐药性或毒性,改变基因的功能(可以被批准的或研究用的药物靶向),则该变异可以视为影响临床护理的生物标志物,可以作为临床试验的纳入标准,影响疾病的预后,帮助确定癌症的诊断或保证实施监视措施以早期发现癌症。因此,临床影响应包括治疗,预后,诊断和预防措施。给定变异的临床影响应根据当前可获得的证据确定。基于变异分类的证据在临床决策中的重要性,可以对其进行不同的权衡。在文献综述和工作组共识的基础上,我们建议将临床和实验证据分为四个级别
- Level A, biomarkers that predict response or resistance to
US FDA-approved therapies for a specific type of tumor
or have been included in professional guidelines as
therapeutic, diagnostic, and/or prognostic biomarkers for
specific types of tumors; - Level B, biomarkers that predict response or resistance to
a therapy based on well-powered studies with consensus
from experts in the field, or have diagnostic and/or
prognostic significance of certain diseases based on wellpowered
studies with expert consensus; - Level C, biomarkers that predict response or resistance to
therapies approved by FDA or professional societies for a
different tumor type (ie, off-label use of a drug), serve as
inclusion criteria for clinical trials, or have diagnostic
and/or prognostic significance based on the results of
multiple small studies; - Level D, biomarkers that show plausible therapeutic
significance based on preclinical studies, or may assist
disease diagnosis and/or prognosis themselves or along
with other biomarkers based on small studies or multiple
case reports with no consensus.
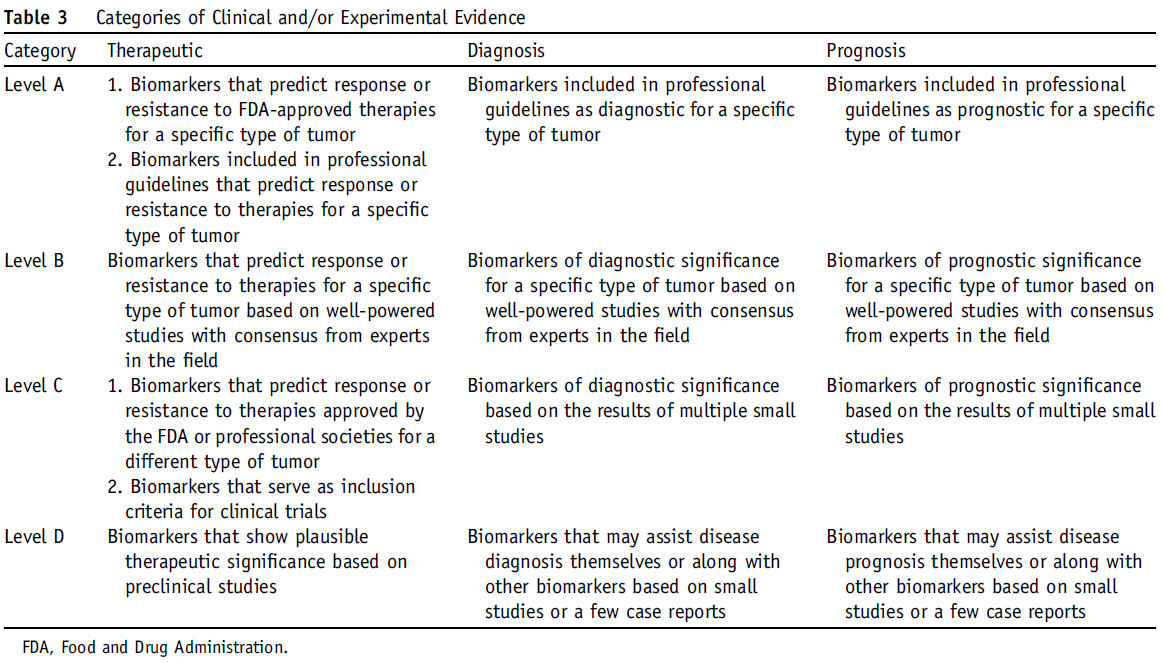
可以将这些证据水平分配给基因组变异体,以确定其临床影响的重要性。 我们建议根据体细胞疾病的临床影响将其序列变体分为四类:
- tier I,具有强烈临床意义的变体(A和B级证据);
- tier II,具有潜在临床意义的变体(C或D级证据);
- tier III,临床意义未知的变体;
- tier IV,良性或可能良性的变体
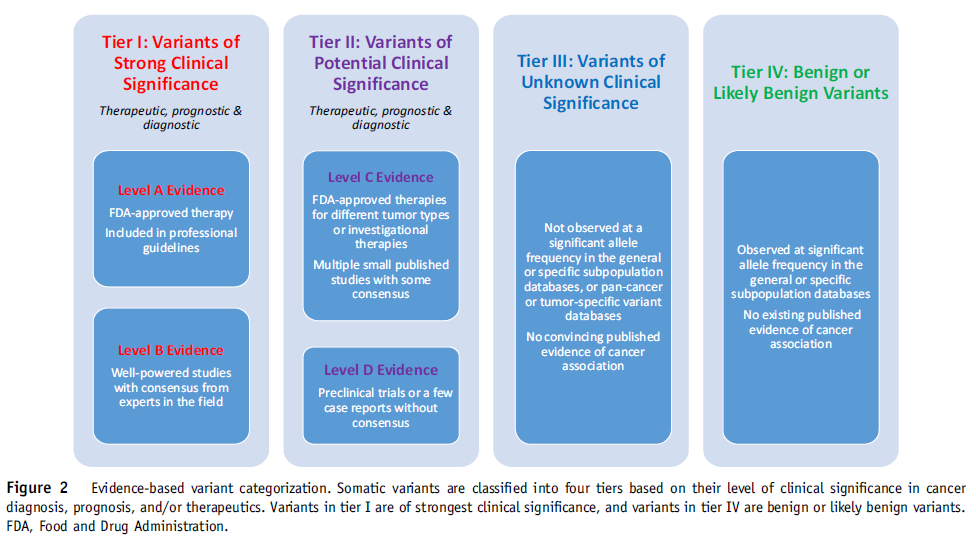
各类别判断依据
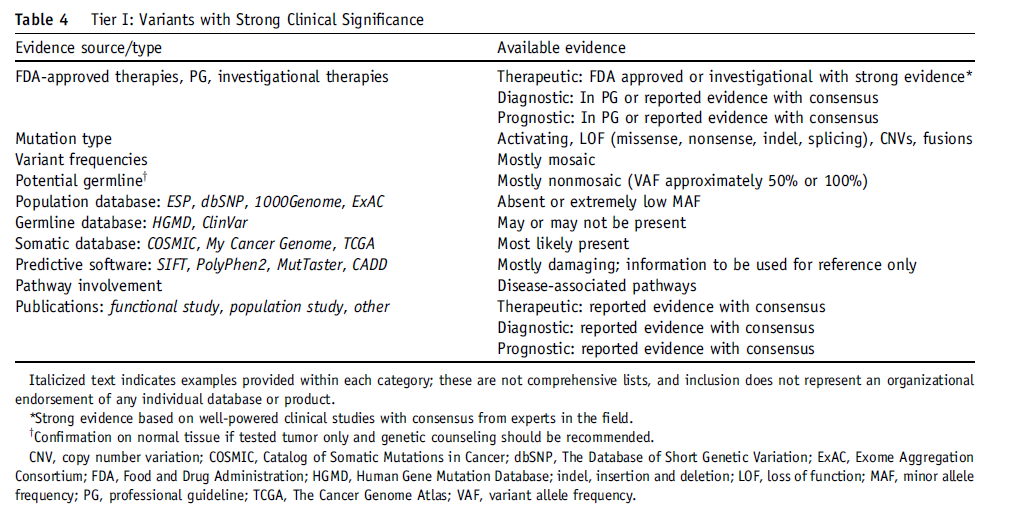
Tier I Variants: Variants with Strong Clinical Significance (Level A and B Evidence)
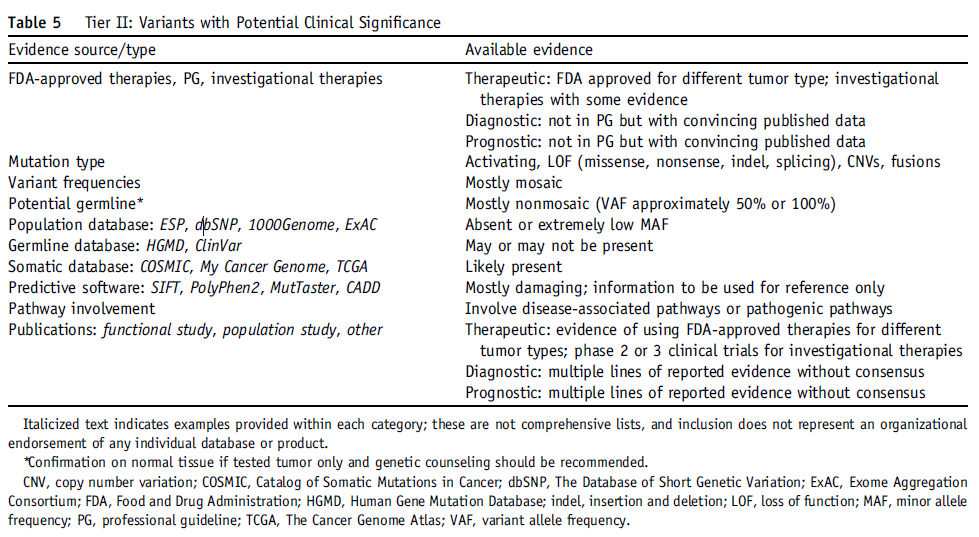
Tier II Variants: Variants with Potential Clinical Significance (Level C and D Evidence)
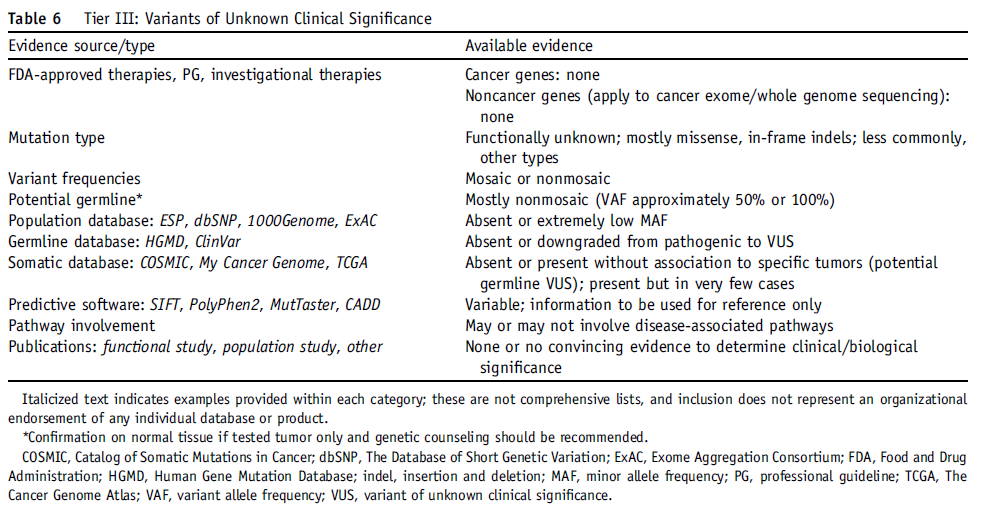
Tier III Variants: Variants of Unknown Significance
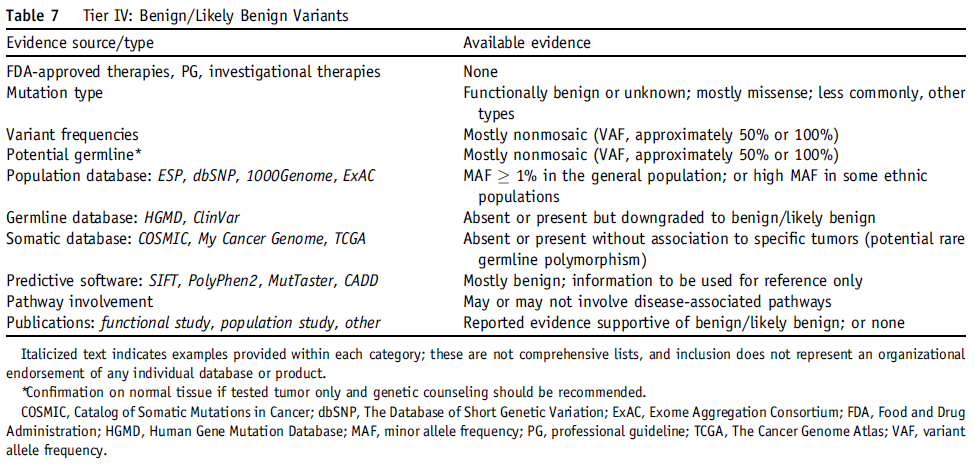
Tier IV Variants: Benign or Likely Benign
Germline Variants Identified during Cancer Testing
在体细胞致癌突变的临床实验室研究中,重要的是将获得的体细胞变异体与遗传的胚系变异体区分开。大多数胚系变异体都是遗传变异,这些变异可以代代相传,并且通常在100%的细胞中具有变异,导致等位基因分数为0.5或1.0。体细胞变异是在出生后获得的,通常是由于DNA复制或修复错误或环境侵害引起的。由于存在污染正常组织的情况,因此即使在表面上纯净的肿瘤样本中,体细胞变异的等位基因分数通常<0.5。实验室必须正确识别可能与诊断,预后,治疗干预和/或临床试验选择有关的体细胞突变,并且不得将高频体细胞突变错误识别为胚系变异,因为这可能具有重大的临床意义。同样,实验室必须认识到可能导致癌症易感综合症的胚系变异,这将对患者和其他家庭成员产生医疗保健影响。
为了帮助对变异进行分类,一些实验室在对正常的,匹配的对照DNA样品进行测序的同时,对肿瘤DNA进行了平行测试。当正常,匹配的对照组织与肿瘤一起测序时,胚系变异通常很明显。 In this case, the laboratory must have policies that address detection, disclosure/nondisclosure, and interpretation/reporting of germline variants (see section below). 当分析中未包括匹配的对照时,实验室应具有可用于推断变体是体细胞或胚系的标准。指定胚系的主要标准是VAF,对于杂合变体,应为约50%,对于纯合变体,应为100%。某些种系变体(例如大插入缺失)可能会导致正常等位基因的优先扩增(在基于扩增子的测试中)或捕获(在基于捕获的测试中),因为这些等位基因的序列同源性丧失,导致<50%用于种系变体的VAF。当在已知的癌症易感综合征基因(例如TP53或BRCA1)中检测到明显的胚系变异时,有关疾病发作年龄的临床信息(年轻人与致癌基因中遗传的种系突变的较高风险相关) ,肿瘤的侧面性(更可能遗传双侧肿瘤)以及癌症的家族或个人病史可以帮助确定癌症易感性的可能性。文献综述和数据库查询还可以帮助确定该变异体先前是否已被报告为易感综合征患者的复发种系变异体。构成突变的有用数据库包括:
- Online Mendelian Inheritance in Man (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/ omim, last accessed March 6, 2016),
- Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php, last accessed March 6, 2016),
- ClinVar
- locus-specific databases
实验室应制定一项政策,对恶性肿瘤中发现的遗传变异进行测试,以在收到患者的适当同意或根据临床医生的要求后,使用经过临床验证的生殖测试来确认变异的生殖或体细胞来源。 ACMG建议在胚系测试中显示阳性结果,以显示53个基因的阳性结果,其中大约一半与可能在体细胞检测面板上的癌症易感性基因有关。即使在仅将该胚系作为一部分进行评估的情况下,也建议进行公开 肿瘤/正常研究。 通过推断,在仅肿瘤的体细胞突变研究中也考虑种系致病变异的可能性似乎是谨慎的。
在单样本无法判断变异是体系还是胚系变异是,添加局限性说明
When paired germline samples are not used, NGS analysis
does not distinguish germline and somatic variants, and
sequencing results may contain both findings. In this case,
findings can be reported with a disclaimer that the NGS test
used does not allow definitive differentiation between
germline and somatic variants. In certain settings, a germline
variant may be suspected (eg, MAF 40% to 60%). However,
this interpretation should be made with caution and correlated
with tumor cellularity. If a germline variant is suspected,
testing of a patient germline sample (eg, blood in patients
with solid tumors) can be suggested if clinically indicated
after an appropriate patient consent. The reports should
include a statement addressing the manner in which the
distinction between somatic and germline alterations is made,
and indications of remaining uncertainty, where appropriate.
“
Interpretation and Reporting
Nomenclature
所有检测到的遗传变异均应按照HUGO基因命名委员会的指定进行注释和报告:
- 基因名参考 :HGNC :http://www.genenames.org
- 变异写法参考:HGVS,http://www.hgvs.org
SNV和插入缺失应使用p.和c. 符号进行报告(例如,BRAFp.V600E,c.1799T> A)。
SV,列出两个融合的基因伴侣之间用斜杠分隔(例如,EML4 / ALK fusion)。
CNV应以表格形式报告为拷贝数GAIN或LOSS。
如果适用,可以包括基因/基因组基因座的基因组坐标。
可以在适当的时候报告数字拷贝数的变化[例如,EGFR拷贝数GAIN(拷贝数比25); CDKN2A拷贝数LOSS]。
使用标准术语不会超过与临床团队进行清晰明确沟通的需要。 根据需要,除了标准术语外,还应包括口语命名法,以便医生阅读报告并使用它们来确定治疗时向医生清晰地传达含义。例如,TERT启动子变体的报告可以为1-124C> T( HGVS命名法,然后是括号中的口语命名法(TERTC228T)。使用HGVS命名法报告基因组变体以明确将变体重新映射到参考基因组,而口语命名法则向临床团队传达了明确的信息。
Other Reporting Elements
除了检测到的变体之外,报告还应包含其他一些元素,这些元素可能与更深入的结果分析或与随时间推移从该患者获得的其他结果进行比较有关,例如基因组座标,基因组构建和转录参考序列( 例如NM_004333.4),前提是此信息不会损害患者和临床提供者解释该报告中与之直接相关的基本要素的能力。
本节或对结果进行扩展说明的另一节中,远离顶部结果。应评估等位基因分数(VAF)和覆盖率,并在适当时包括在报告中。该报告应包括所用NGS测定的测序覆盖率临界值。在报告中应声明所有未满足最低要求的测序覆盖率标准的基因和/或热点均已失败。
报告不应仅限于肯定的发现。 I类药物/癌症组合应包括相关的阴性结果(例如,肺癌患者中明确缺乏EGFR突变或黑色素瘤患者中明确缺乏BRAF突变)。如果存在不确定性, 必须在报告中进行沟通;这包括序列质量,样品充分性,肿瘤含量和生物医学知识的问题。
Reporting of Germline Variants
希望对配对的胚系样品进行并发分析,因为这可以澄清解释。但是,这并不总是实用的,因此也不是必需的。当有成对的胚系样品可用时,测序管线可允许从体细胞获得性变体中分离出胚系发现。通常,仅解释和报告体细胞变异。如果患者或临床医生要求对胚系发现进行进一步分析,则可以在以后重新查看胚系NGS数据,并在获得患者适当同意后进行报告。成对的胚系测试可能需要获得同意并提供相关文件,以符合当地法律和政策。如果NGS小组的某些基因中未报告胚系变体,则初始报告应特别说明这一事实。当不使用成对的胚系样品时,NGS分析不能区分胚系和体细胞变体,测序结果可能包含这两个发现。在这种情况下,可以用免责声明报告使用的NGS测试不能明确区分胚系和体细胞变体。在某些情况下,可能会怀疑胚系变异(例如MAF 40%至60%)。然而,这种解释应谨慎进行,并与肿瘤细胞数量有关。如果怀疑胚系变异,如果在适当的患者同意后临床上有指示,可以建议对患者胚系样本(例如,实体瘤患者的血液)进行测试。报告中应包含一份声明,说明在体细胞和胚系变化之间进行区分的方式,并在适当情况下指示仍存在不确定性。首先测试癌症样品和稍后再配对的胚系样品的实验室可以选择进行以下操作:
- 将包含胚系样品发现的结果发布到癌症报告的附录中,
- 在胚系报告中单独发表一份关于胚系变体的报告,并附上唯一的解释性声明,并在初始癌症报告中添加一份附录。
- 作为单独的胚系报告和另外的单独报告发布,该报告整合了癌症和胚系样品的结果。
生殖细胞变体应按照ACMG / AMP指南进行报告。如果订购了针对癌症易感基因的生殖细胞测试,则生殖细胞变异的报告应遵循ACMG / AMP指南。应提供遗传咨询和转诊给临床医学遗传学家。实验室应制定政策,以报告重要性不明的变体并披露次要发现,包括在何种情况下将报告或不报告此类发现
Reporting the Clinical Significance of DetectedVariants
对检测到的遗传变异提供解释性注释是很有用的,该变异将这种变异置于临床病理背景下可为管理决策提供依据。这对于I级和II级突变至关重要。必须对III级变体的详细分析与目标,使报告中最关键的信息保持简洁,清晰并突出显示的目的相平衡。这些评论可能包括该变体对于特定肿瘤类型,对生化途径的影响以及相关癌症的患病率的功能,预后或预测意义。但是,建议应该简短明了,并应谨慎措辞,并应理解治疗或其他患者管理决定是基于除遗传改变以外的许多医学信息,其中许多信息对于分子专业报告是不可用的。重要的是要认识到,治疗的适用性是基于许多因素,而不是根据测试申请书上写的诊断和通过测试发现的基因型。这些因素通常是分子专业报告结果所未知的(即,存在混杂的医学状况,例如葡萄糖耐量不足,自身免疫性疾病或心力衰竭),并且在推荐特定疗法时未考虑这些其他因素会导致混淆,患者与肿瘤科团队之间的冲突以及焦虑。 NGS实验室报告中的治疗建议应以证据为依据,应与患者的癌症诊断相关,并应使用某种语言来明确指出该报告包含结合实验室可用数据点的广义治疗建议(例如,diag-疾病和基因型),但需要将其他因素纳入为每个个体制定治疗计划的过程中。尽管可以接受有关相关试验的一般性陈述或已发表试验的引用结果,但不应针对具体临床试验提出建议。
网上查找的基于ACMG 构建的思维导图,可以作为参考资料