摘要简介
图形总结
cfDNA BIOLOGY
- cfDNA主要通过细胞凋亡和坏死释放,也可能通过活性分泌释放。
- cfDNA的半衰期为16分钟至2.5小时。
- 在体内,cfDNA要么被核酸酶清除,然后被肾脏排泄到尿液中,要么被肝脏和脾脏吸收,随后被巨噬细胞降解。
- 体外分离cfDNA的保存温度不超过 -20 ℃,且不超过1次冻融循环,以防止cfDNA降解。
- cfDNA片段的稳定性可能通过与细胞膜、细胞外囊泡或蛋白质的结合而提高。
- cfDNA在正常情况下浓度通常较低(在100 ng/mL以内),但在某些生理和病理条件下,如运动、炎症、糖尿病、、癌症等,cfDNA浓度会显著升高。健康个体中,cfDNA主要来自淋巴细胞,而在癌症患者中,来自癌症组织的cfDNA含量显著增加。
- 采用基于测序的方法测定cfDNA时,166bp的峰值显著,而肿瘤来源的cfDNA长度(约为144 bp)较正常cfDNA较短。
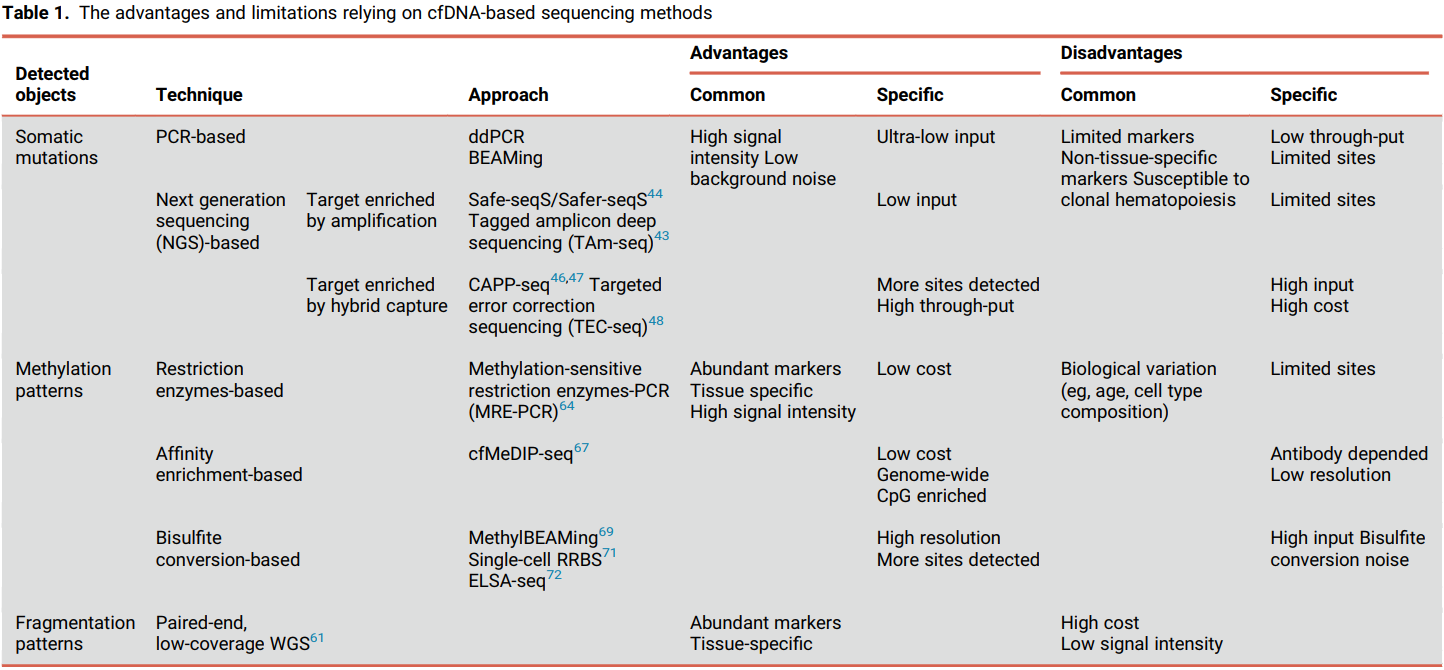
目前基于cfDNA进行肿瘤早筛的测序方法
癌症早期检测的策略通常是基于检测癌细胞释放的cfDNA中与癌症相关的改变,即 ctDNA(circulating tumor DNA)。血浆中ctDNA的浓度较低,占cfDNA总浓度的不到0.01%,尤其是在早期癌症中,对癌症的早期检测提出了很大的挑战。目前,突变、甲基化和片段模式是cfDNA早期检测癌症的主要测序生物标志物。
Mutation-based sequencing approach and strategy
基于突变的测序方法在早期癌症检测中的理想应用需要较低的检测下限(LOD)来区分在DNA提取、文库制备、靶标富集、杂交过程和测序本身过程中可能引入的背景噪声(如假阳性),这使得检测真正的突变更加困难。一些背景噪声可以减轻,如在提取过程中氧化应激损伤引起的序列改变,碎片化和杂交过程。为了提高目标测序的分析灵敏度,独特分子标识符(uniquemolecular identifiers, UMIs),即直接连接在文库DNA片段两端的独特分子条形码,被用于促进同一DNA片段序列的生物信息学比对。这种UMI策略可以帮助减少后续分析中的人为错误。深度测序(CAPP-Seq)方法结合double-umi进行癌症个性化分析时,cfDNA的LOD可达0.02%。
基于cfDNA突变的分析有几个局限性:
- 靶向测序的芯片设计主要基于现有的关于癌症基因改变的知识。
- 假阴性结果不仅在癌变突变超出定制检测范围时不可避免,而且血浆ctDNA浓度不足也可能导致假阴性结果。
- ctDNA在晚期和转移癌患者中的浓度高于早期癌症患者,且随癌症类型的不同而不同。它在肝癌、胆道癌、食道癌和卵巢癌中的含量也较高,但在前列腺癌、乳腺癌和结肠癌中的含量相对较低此外,一些因素可能影响cfDNA浓度的变化,如年龄、体重指数、和生理参数基于突变的分析也受限于肿瘤肿块中最初出现的突变数量,这因癌症而异。黑色素瘤和肺癌平均有超过8.9个突变/Mbp,而低级别胶质瘤、乳腺癌、胰腺癌和前列腺癌平均有少于2.2个突变/Mbp此外,早期癌症患者的肿瘤突变负担比晚期癌症患者低,这使得利用基于突变的技术检测癌症更加困难。
- 假阳性结果可能发生在大量未患癌症的个体中,由于克隆造血(染色质免疫沉淀[CHIP])相关突变。在来自NSCLC和健康对照的cfDNA中检测到的变异中,58.0%和90.0%也在匹配的白细胞(WBC)中检测到,强调了对匹配的WBC DNA应用等效测序深度以排除CHIP干扰的重要性。
Methylation-based sequencing approach
基于表观遗传改变的cfDNA测序方法被认为是有希望的癌症早期检测的替代方法。CpG位点甲基化是基因表达、组织分化、器官发育、衰老和肿瘤发生的重要表观遗传学调控机制。肿瘤特异性DNA甲基化的变化发生在肿瘤发生的早期,有时甚至在基因突变发生之前。先前的一项研究表明,在临床癌症诊断前四年,血浆中就检测到了甲基化的变化。此外,与只发生在少数基因组位置的典型癌症突变不同,近3000万个甲基化位点分布在人类基因组中,使它们无处不在,成为癌症检测的丰富信号。值得注意的是,甲基化模式通常是组织特异性的,这使得TOO(tissue of origin)分析成为可能。
全基因组亚硫酸氢盐测序(WGBS:Whole genomic bisulfite sequencing)是获取全基因组DNA甲基化图谱的金标准。然而,然而高昂的成本,输入DNA回收率低,测序深度有限,使其无法在临床应用。
基于二代测序(NGS)的甲基化测序方法由于成本较低、测序深度较高,受到越来越多的关注。基于ngs的方法包括亚硫酸氢盐预处理(还原亚硫酸氢盐测序[RRBS])、酶切(甲基化敏感限制性内切酶测序)和亲和富集(甲基化DNA免疫沉淀和高通量测序)。亚硫酸氢盐转化后的DNA回收率低也是基于ngs方法的一个问题。cfMeDIP-Seq (cfMeDIP-Seq)是一种基于免疫沉淀的协议,采用传统的MeDIP-Seq,允许在无亚硫酸氢盐的过程中,低输入DNA的全基因组甲基化分析。
与需要100 - 2000 ng DNA输入的MeDIP-Seq、RRBS和WGBS相比,cfMeDIP-Seq在观察到的和预期的差异甲基化区域数量之间产生了近乎完美的线性关联(r2 = 0.99, p <0.0001),只有1 - 10ng的DNA输入。
近年来的技术进步,如methylBEAMing、单细胞RRBS和增强的线性分裂扩增测序(ELSA-Seq),通过减少所需的DNA输入量和提高分析灵敏度来促进cfDNA甲基化测序的应用。例如,ELSA-Seq构建了一个单链文库,其甲基化覆盖深度高,扩增偏置小,可重复性极高,输入量可达500 pg。此外,相邻的CpG位点通常代表共甲基化状态,将多个基因组距离近、相关性高的CpG位点整合为甲基化单倍型区块,可进一步提高检测精度。
cfdna甲基化分析也有局限性:
- 癌症中存在的表观遗传改变也可能存在于其他非癌组织中,这可能导致假阳性。例如,食道癌和巴雷特食管有几个相似的甲基化改变。此外,
- 甲基化改变随着年龄的增长而积累,大约5%的CpG位点表现出与衰老和肿瘤发生共同的显著变化。同时,
- 当检测信号低于LOD时,可能会出现假阴性结果。同样,cfDNA的检出率与cfDNA的浓度有完整的相关性,而cfDNA的浓度也受癌症病理类型的影响。此外,这些
- 基于甲基化的测序方法可能无法检测主要由基因突变、体细胞拷贝数畸变或融合事件驱动的癌症,例如主要由EGFRL858R突变、EML4-ALK融合或ERBB2扩增驱动的几种肺癌亚型。
cfDNA fragmentation-based sequencing approach
cfDNA的全基因组片段化为癌症的早期检测开辟了新的领域。全基因组测序(WGS)显示癌源cfDNA片段的长度比非癌源cfDNA片段的长度变化更大。cfDNA片段模式的差异反映了癌症中染色质结构的变化,以及其他基因组和表观基因组异常。最近,一种结合全基因组片段化的机器学习模型可以将多个癌症患者与健康对照组区分开来,敏感性从57%到99%以上不等,特异性为98%,并在75%的病例中识别出癌症的TOO。除了片段长度之外,cfDNA片段在基因组中的断裂位点还揭示了核小体占据的全基因组地图,提供了丰富的信号。
局限性:
- 尽管WGS能够从极少量的cfDNA中同时分析数十到数百种肿瘤特异性异常,但其覆盖深度较低,成本较高。
- 基于cfDNA片段的方法与cfDNA甲基化分析有相同的局限性,如生理等病理条件导致的假阳性和技术限制导致的假阴性。
三种技术方法的整体比较