变异检测
变异过滤
一些可以获取的临床变异检测资料信息
中检院TMB标准化项目
采用多个变异检测软件进行交叉验证,而后进行人工审核确认。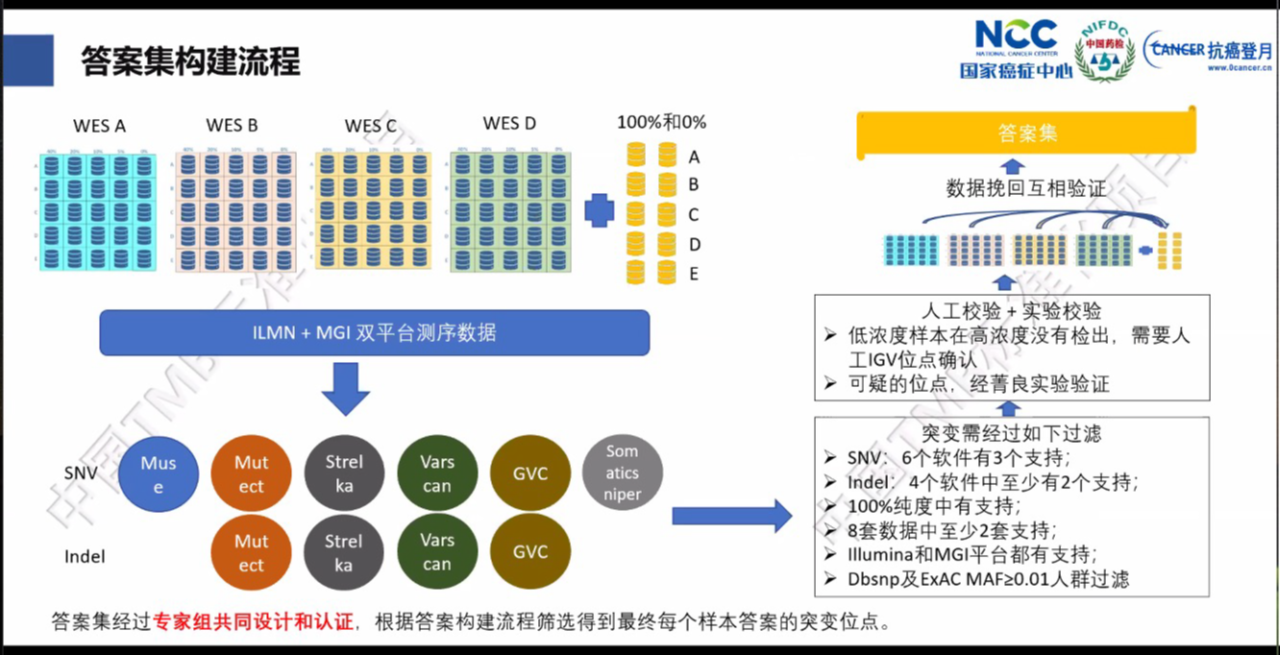
全国实体肿瘤体细胞突变 高通量测序(大 Panel)检测
借助质评自身的特殊性,通过多家检测机构的结果进行较差验证,按着特定标准筛选获得标准答案集合。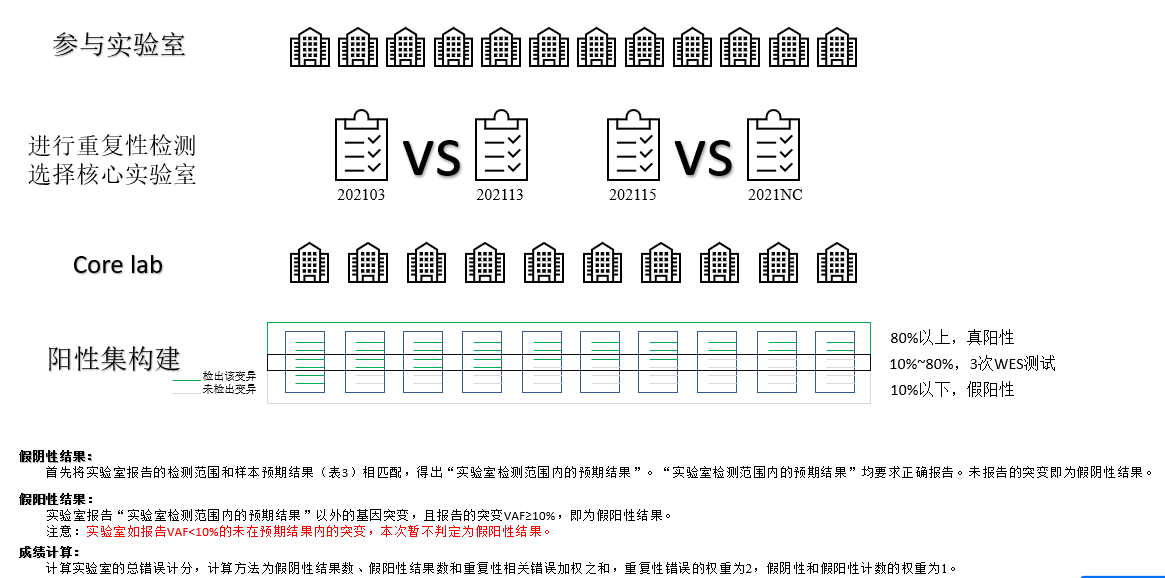
基于IGV的人工审核
针对如果对变异结果进行人工审核,2019年有一篇发表在GENETICS IN MEDICINE (IF 9.108)的文章,可以为我们提供一些参考。Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples
其中针对突变审核包含如下过程:
步骤1:把突变可视化
使用IGV观察突变;确保IGV与插件中突变选取保持一致;通过插件的“S”使突变发生reads置顶;确认IGV、插件的reads展示一致;步骤2:确认支持突变的数量
查看突变的①链方向、总覆盖度、②位点的突变频率、③非突变频率;考虑突变受到的其他因素影响,比如原发性肿瘤 DNA、复发性DNA、肿瘤RNA等;步骤3:确认支持突变的质量
寻找多个不匹配或者与ref高度差异性的区域;寻找半透明或透明的reads或碱基(比对质量低的);①对有疑问reads进一步确认比对质量、碱基质量;②看突变对应的normal检出情况;
步骤4:检查测序误差
①切换“成对查看”确认短插入片段情况;②IGV缩小确认是否在高度差异性区域,临近区域是否有indel存在;③看参考序列是否在低复杂性区域,是否存在串列重复序列;步骤5:给突变选择符合哪个Call标签
通过突变质量、突变数量的信息,选择合适的Call标签;步骤6:给突变选择符合哪个Tag标签
可以对每个突变进行tag标签标记,对Call标记 不确定、失败的突变,尤其重要;步骤7:给突变写附加的备注信息
其中提供了19中tag,作为变异过滤过程中参考判断的指标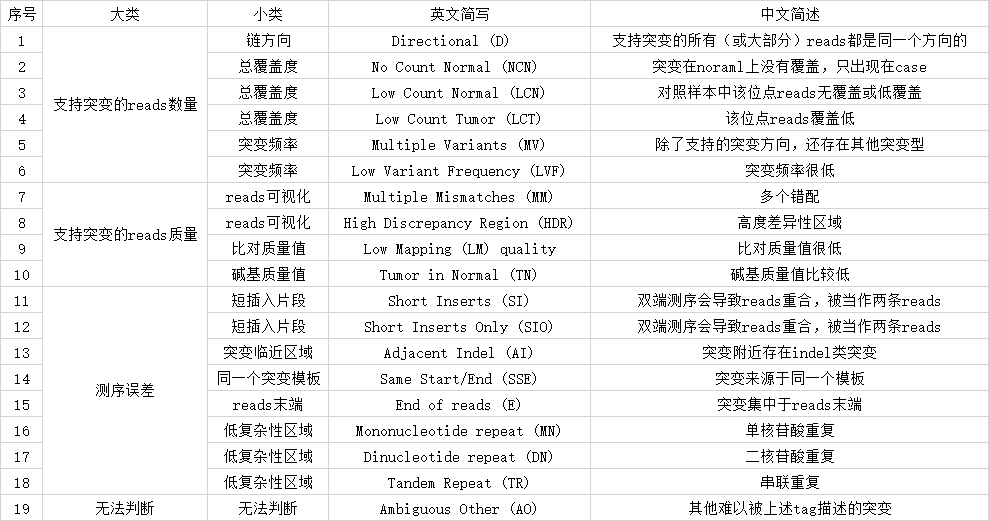
燃石检测流程
实际比较难获得泛生子内部资料,只能借助一些发布文章的method,这也往往是一个非常有效的办法。
在燃石的官网查到燃石的学术发表
找到其中一篇文章从题目看是和NGS检测为主的Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology
从文章的method中,我们可以看到泛生子流程的整体检测逻辑如下:
制作模拟数据
- 通过wgsim (v1.9) 进行数据模拟
变异的分析流程
- 首先对下机数据进行Trim TrimGalore
- 使用anaquin 进行sequin(spike-in controls)分析, 将sequin的Reads进行分离,然后矫正到样本相同的深度,并通过Anaquin somatic 进行变异检测。详细见参考文献Use of synthetic DNA spike-in controls (sequins) for human genome sequencing
- bwa mem (v0.7.16) 进行数据比对,比对到Hg38
- 剔除捕获区间外的Reads,并用gatk MarkDuplicates (v4.0). 标记重复
- VarScan (v2.4.3) 用来进行 SNVs and indels 的检测(最少支持数是3条read-fragments)
泛生子检测流程介绍
在泛生子官网查找泛生子的成果,
2016:The genome-wide mutational landscape of pituitary adenomas的附件中提到了分析流程
2019:Detection of early-stage hepatocellular carcinoma in asymptomatic HBsAg-seropositive individuals by liquid biopsy
详细分析方法参考
文档中提及的方法材料:
- Sequencing reads were primarily processed with our own program to extract tags and remove sequence adapters.
- Residual adapters and low-quality regions were subsequently removed using Trimmomatic (v0.36).
- The cleaned reads were mapped to the hg19 and HBV genomes using ‘bwa(v0.7.10) mem’ with the default parameters.
- Candidate somatic mutations, consisting of SNP and INDEL, were identified using samtools mpileup (9) across the targeted regions of interest.
- To ensure accuracy, reads with the same tags, and start and end coordinates were grouped into Unique Identifier families (UID families). UID families containing at least two reads and in which at least 80% of reads were the same type were defined as Effective Unique Identifier families (EUID families). Each mutation frequency was calculated by dividing the number of alternative EUID families by the sum of alternative and reference ones.
- The mutations were further manually reviewed in IGV.
- The candidate variations were annotated with Ensembl Variant Effect Predictor (VEP) (10). HBV integrations were identified using Crest (11) , and at least 4 soft-clip reads supports were needed.
世和
Sequencing and data processing
- Target enriched libraries were sequenced on the HiSeq4000 platform (Illumina) with 2×150bp pair-end reads.
- Sequencing data were demultiplexed by bcl2fastq (v2.19),
- analyzed by Trimmomatic 24 to remove low-quality (quality<15) or N bases,
- mapped to the reference hg19 genome (Human Genome version 19) using the Burrows-Wheeler Aligner.
- PCR duplicates were removed by Picard.
- The Genome Analysis Toolkit (GATK) was used to perform local realignments around indels and base quality reassurance.
- SNPs and indels were called by VarScan2 and HaplotypeCaller/UnifiedGenotyper in GATK, with the mutant allele frequency (MAF) cutoff as 0.5% for tissue samples, 0.1% for liquid biopsy samples, and a minimum of three unique mutant reads.
- Common variants were removed using dbSNP and the 1000 Genome project. Germline mutations were filtered out by comparing to patient’s whole blood controls.
- The resulting somatic variants were further filtered through an in-house list of recurrent sequencing errors that was generated from over 10,000 normal control samples on the same sequencing platform.
- Gene fusions were identified by FACTERA.
- copy number variations (CNVs) were analyzed with ADTEx . The log2 ratio cut-off for copy number gain was defined as 2.0 for tissue samples and 1.6 for liquid biopsy samples. A log2 ratio cut-off of 0.67 was used for copy number loss detection in all sample types. The thresholds were determined from previous assay validation using the absolute CNVs detected by droplet digital PCR (ddPCR). Allele-specific CNVs were analyzed by FACETS 30 with a 0.2 drift cut-off for unstable joint segments. The proportion of chromosomal instability (CIN) was calculated by dividing the size of drifted segments by the total segment size.
- Tumor mutational burden (TMB) in this study was defined as the number of somatic synonymous mutations per megabase in each sample, with hotspot/fusion mutations excluded.
贝瑞
In brief, DNA extracted from FFPE tissue biopsies was fragmented to an average size of 300 bp, molecules were then end repaired and A-tailed and finally T tailed linkers were ligated on. The added linkers were a mix of 96 different molecular barcodes giving a high probability that each molecule was marked differently at both ends and thus uniquely barcoded. Libraries were amplified by PCR and resulting amplicons captured using biotinylated probes (120 nucleotides) for the 457 genes. Following elution, molecules were re-amplified using complementary sequencing primers and then paired end (PE) sequenced (2 × 150 bp) on the NovaSeq platform (Illumina).
- Fastq sequencing reads were aligned to the hg19 reference genome using the Burrows Wheeler algorithm (Li and Durbin 2009).
- The resulting SAM files were converted to BAM file format and then sorted on genome coordinates using Samtools.
- To remove PCR bias (reads with the same molecular barcodes and same start and same stop positions), only the unique coded molecules were used for copy number analysis.
- After filtering out low mapping quality reads (MAQ < 20), the average depth of coverage (DoC) for each target was calculated using the GATK Depth Of Coverage algorithm (McKenna et al. 2010).
- After GC correction using LOESS regression method (Alkan et al. 2009), reads were normalized using the RPKM method (Chiang et al. 2019). For these steps, the tumor and matched normal sample was processed separately.
- Somatic SNVs and indels were finally identified by MutLoc (Berry Genomics in-house tools, unpublished), which maps the alternative base fraction compared to the hg19 reference genome.