1. 引言与问题背景
给定一条目标三维骨架(通常以主链坐标表示),蛋白质序列设计(protein sequence design)要回答的问题是:应填入怎样的氨基酸序列,才能使该序列在物理上稳定地折叠成(或组装成)所给定的结构形态。它与从序列预测结构的结构预测方向相反,因此常被称为逆向折叠(inverse folding)。在深度学习兴起之前,大量可实验验证的从头(de novo)设计依赖 Rosetta 等基于物理能量与侧链 rotamer(离散侧链构象库)搜索的方法;而结构预测领域已被 AlphaFold 等模型深刻改写之后,序列设计同样需要兼顾速度、可扩展性、多链与对称约束以及对不完美骨架的鲁棒性。
ProteinMPNN(Protein Message Passing Neural Network)由 Dauparas 等发表于 Science(2022),由华盛顿大学蛋白质设计研究所(Institute for Protein Design)等团队提出,是一套面向广泛设计任务的深度序列设计框架:在保持计算极轻量的同时,在计算机内指标与湿实验上均表现出显著优势。
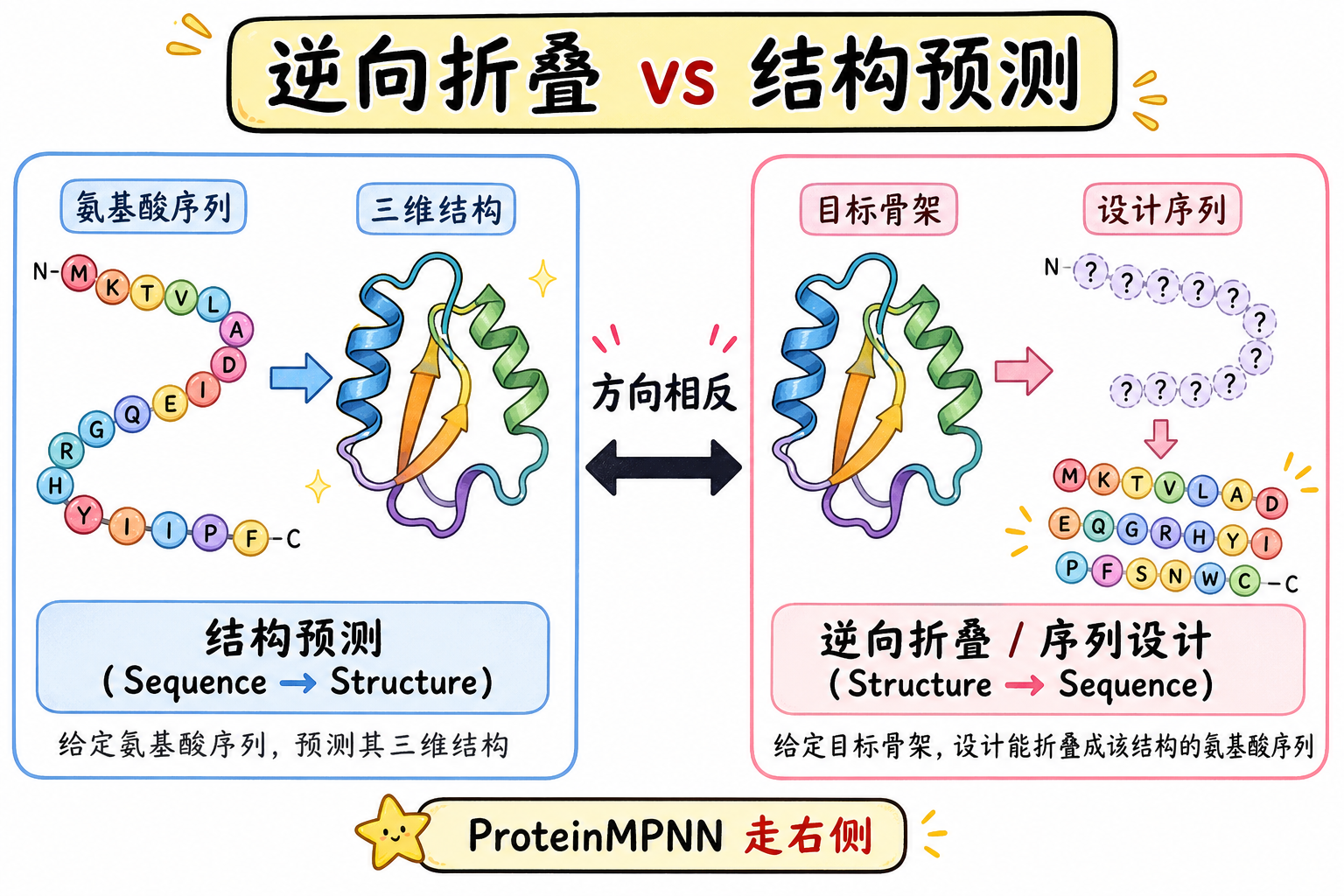
段末注释:逆向折叠指由结构反推序列;从头设计指不直接拷贝天然序列模体的全新拓扑与序列设计。
2. 核心思想:把骨架变成图,把序列生成变成条件建模
ProteinMPNN 将输入结构视为一张图:节点为残基,边由几何近邻(例如按 Cα 的 k 近邻)定义。模型不直接枚举侧链构象,而是利用主链提供的几何与化学环境线索,学习从「局部邻域几何 → 该位点氨基酸类型」的映射;再通过自回归(autoregressive)方式逐位生成整条序列。这样把传统上昂贵的组合侧链搜索,转化为一次前向网络推理中的条件分类问题。
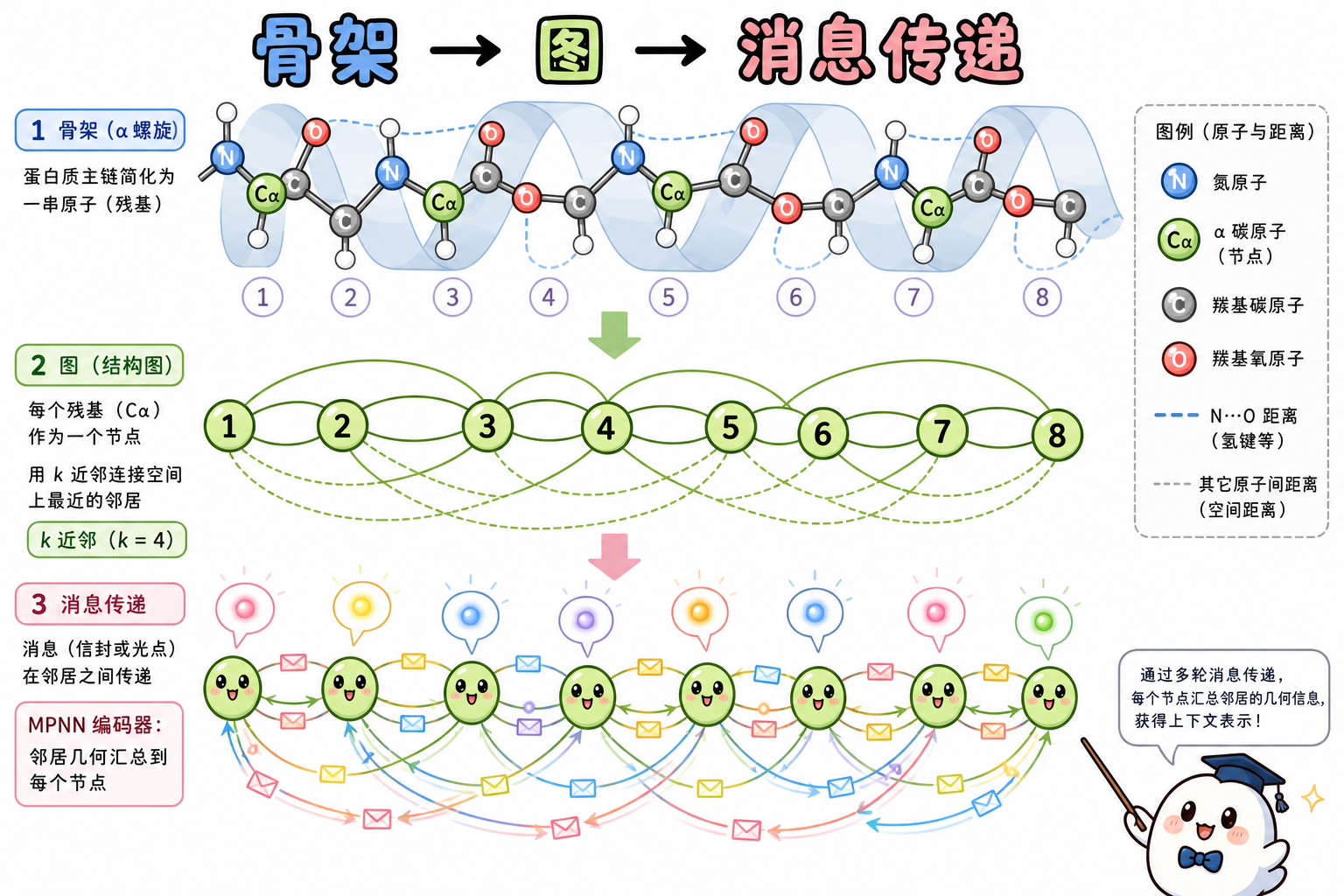
段末注释:自回归指在已生成子序列条件下依次预测下一位置;k 近邻指每个残基在图中连接空间上最近的 k 个 Cα 邻居。
3. 网络架构与输入特征
3.1 编码器:消息传递与边更新
模型骨干为消息传递神经网络(Message Passing Neural Network,MPNN):通过多层节点与边的信息聚合,把局部几何上下文编码为每个残基的隐表示。论文在先前 MPNN 序列设计工作的基础上系统做了消融:在仅主链二面角等特征之外,引入 N、Cα、C、O 以及基于主链重建的虚拟 Cβ 等原子对距离作为边特征,可显著提升在天然骨架上的序列恢复率(sequence recovery,即与真实天然序列一致残基的比例);再在编码器中引入边更新(edge updates),使边表示也可迭代 refinement,与更丰富几何特征组合后进一步带来增益。
段末注释:MPNN 指在图上沿边传递并聚合消息的神经网络范式;序列恢复率衡量模型在固定骨架上“猜回”天然序列的能力,是常用的离线评测指标之一。
3.2 邻域尺度
论文系统扫描了 16、24、32、48、64 等 Cα 近邻数,发现性能在约 32–48 个邻居处趋于饱和。这与「序列–结构映射」主要由局部邻域几何决定的经验一致,也说明全连接式全局注意力并非此任务的必要条件——局部图结构已提供较强归纳偏置。
3.3 解码器:随机解码顺序(order-agnostic)
早期自回归模型常固定从 N 端到 C 端解码。ProteinMPNN 改为在训练时对解码顺序做随机排列采样,使模型在推理时可按任务需求选择顺序。其直接收益包括:
- 固定片段上下文:例如结合子设计(binder design)中靶蛋白序列已知、仅设计结合面周边时,可让已知区先被“解码/给定”,未知区在完整上下文下生成。
- 与对称、重复单元、多状态设计兼容:下文单独说明。
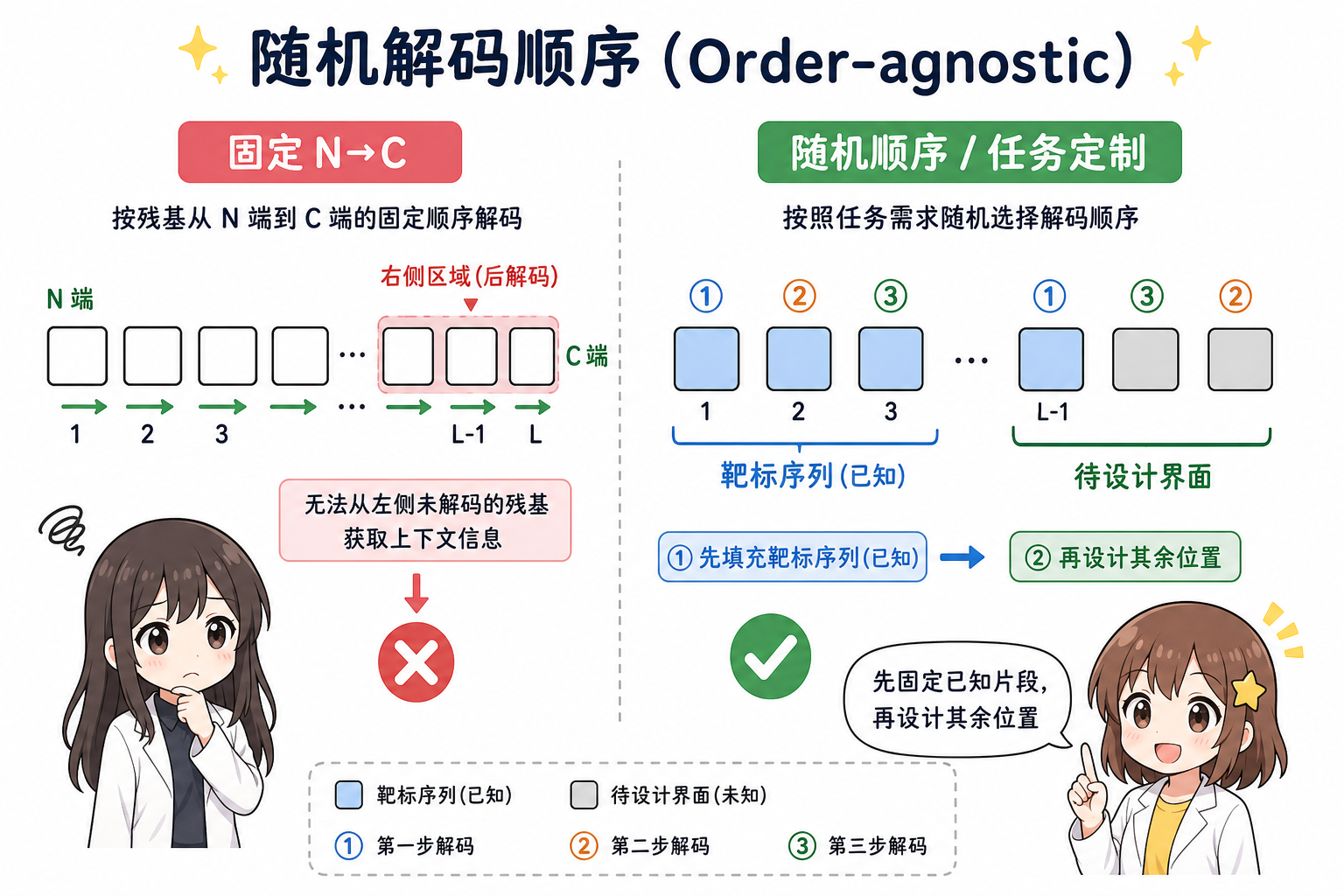
段末注释:结合子指为结合某靶标而设计的蛋白质;固定靶序列、只设计界面属于常见工业与科研场景。
4. 多链、对称与多状态设计
4.1 多链与链序等变
对寡聚体与异源复合物,模型在相对位置编码上保留每条链内部的局域性(论文将链内相对位置编码截断在 ±32 残基量级量级),并加入是否同链的二值特征,以区分链内与链间相互作用。这样在统计意义上对「链的标签顺序」更稳健,适配 PDB 中常见的同源/异源组装。
4.2 位置耦合(tied positions)与对称
对 C2 同源二聚体等情形,需要链 A 与链 B 在对应索引处取相同氨基酸。实现上可对对称相关位置同时给出 logits 并做组合(例如平均 logits 后联合采样),从而在一次推理中强制序列对称性。该机制同样适用于重复蛋白(repeat protein)中重复单元之间的序列 tying,以及同时施加环状对称与单元内重复的更复杂约束。
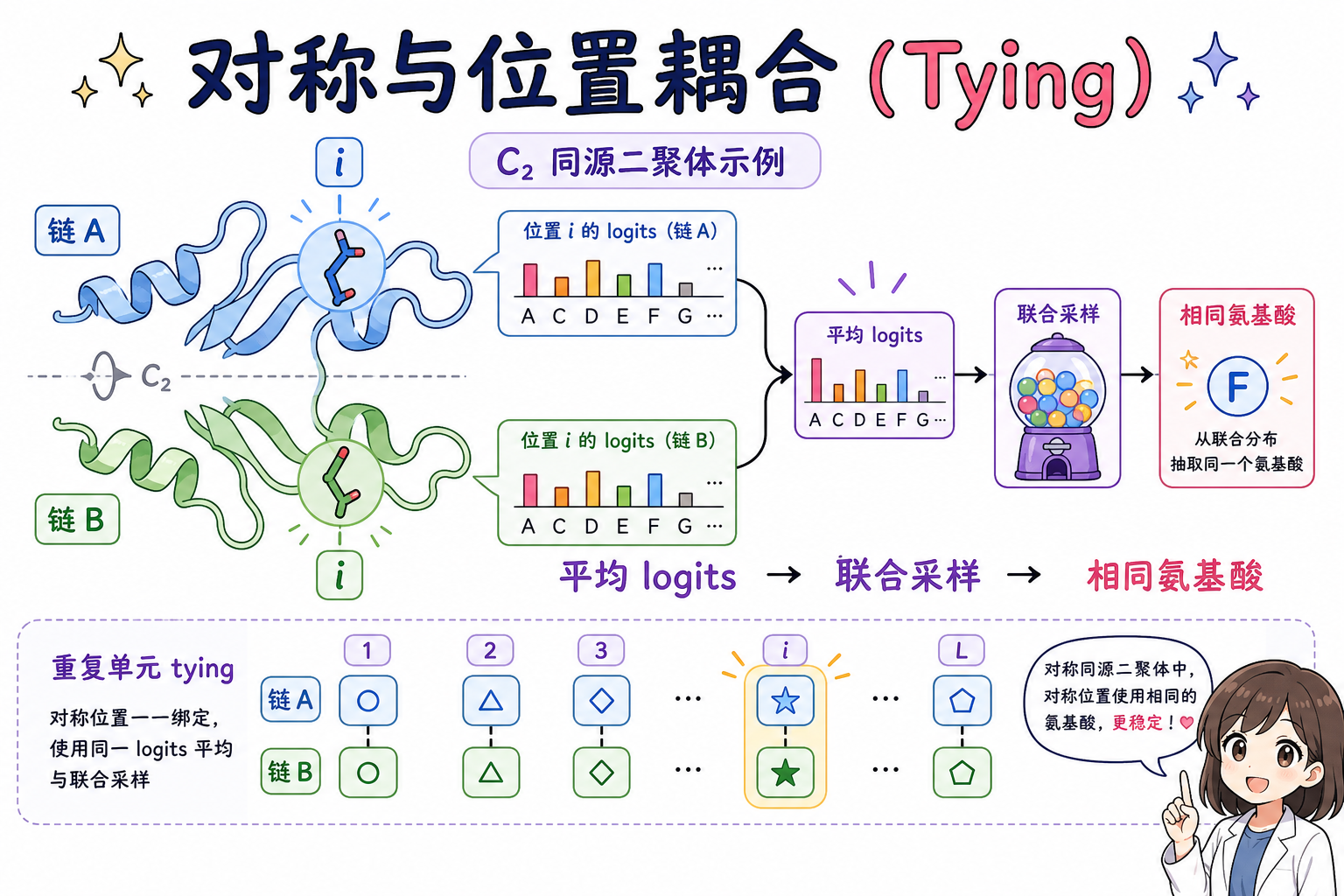
4.3 多状态与显式正负加权
若希望单条序列兼容多个目标构象(multi-state design),可对不同骨架状态分别得到 logits,再做线性组合(系数可为正或负)以同时抬高某些状态、压低另一些状态。该灵活性把序列设计从「对单一静态结构最优」推广到「对一组构象流形或切换路径更鲁棒」。
段末注释:logits 指 softmax 前的非归一化分数;对多状态平均/加权 logits 是一种工程上简洁的多目标折衷。
5. 训练数据与训练噪声
5.1 训练集构建
最终版 ProteinMPNN 使用截至 2021-08-02 的 PDB 中、由 X 射线或冷冻电镜(cryo-electron microscopy,cryo-EM)解析且分辨率优于 3.5 Å、总残基数小于 10000 的组装体;序列按 30% 序列同一性聚类(mmseqs2)以减轻冗余与泄漏,得到约 2.5 万 个聚类代表用于训练。
段末注释:PDB 即蛋白质三维结构公共数据库;聚类可降低同源蛋白过采样带来的虚高指标。
5.2 骨架噪声与“设计友好性”的再定义
仅追求在完美实验骨架上的最高序列恢复,并不等价于最好地服务真实设计流程:实际输入常为预测结构或带误差的模型。论文发现,对主链坐标加入小量高斯噪声(例如标准差 0.02 Å 量级)训练,会在略微牺牲无扰动 PDB 上恢复率的同时,提高在高质量 AlphaFold 模型骨架上的恢复率,并改善后续用 AlphaFold 做单序列结构回验的成功率。
进一步增大噪声(例如至 0.3 Å 量级讨论中)时,模型更关注整体拓扑而非极细微局部几何;在以 lDDT-Cα 等阈值为判据的 AlphaFold 单序列回折评测中,可显著增加高相似度预测的比例——即序列更“强编码”目标骨架。与之配套,推理时可通过温度(temperature)调节采样随机性:较高温度在几乎不显著伤害平均恢复率的前提下提高序列多样性,便于并行湿实验筛选。
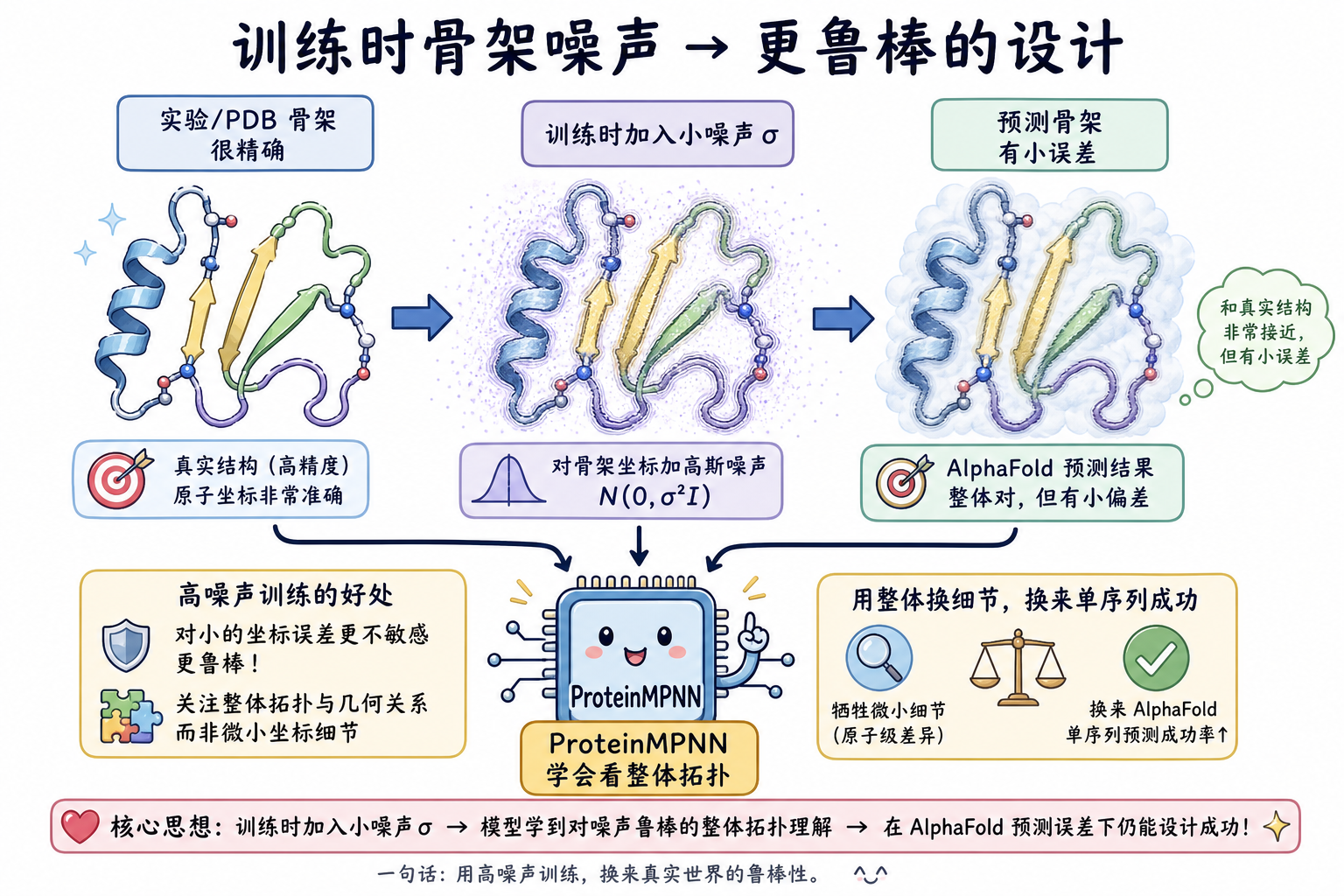
段末注释:AlphaFold 为 DeepMind 等发展的单链结构预测模型族;lDDT(local Distance Difference Test)为局部距离一致性度量,常用于评估预测与参考结构的吻合程度。
6. 计算机内性能要点
- 与 Rosetta 对比:在约 402 条单体测试骨架上,PackRotamersMover 一轮 Rosetta 固定骨架设计与 ProteinMPNN 相比,ProteinMPNN 总体序列恢复约 52.4% 对 32.9%,且在从核心到表面的不同埋藏度区间均更高;计算时间上对 100 残基量级示例约为 1.2 秒 对 4.3 分钟(论文报告的数量级)。
- 寡聚体:在 690 单体、732 同源寡聚、98 异源寡聚测试集上,整体中位数恢复分别约 52%、55%、51%;同源寡聚体中「对称相关位置 logits 平均」优于独立设计或仅平均概率。
- 质量排序:平均对数似然(模型给出的序列在给定结构下的打分)与恢复率在不同温度下呈强相关,可用于快速筛选候选序列。
段末注释:Rosetta 为经典蛋白质建模与设计软件套件;此处对比的是论文设定下的固定骨架组合侧链设计流程,非所有 Rosetta 工作流的普适结论。
7. 湿实验与应用实例(论文内)
下列案例均来自原文系统表征,强调「在固定原设计骨架上仅替换序列」的 rescue(挽救)范式,以突出序列设计方法的边际贡献。
- AlphaFold 幻觉(hallucination)骨架:对 AF 优化得到的非常规拓扑骨架,原始 AF 伴随序列在大肠杆菌中大多难溶;换用 ProteinMPNN 重新赋序列后,可溶性表达与中位产量显著提升,尺寸排阻色谱(Size Exclusion Chromatography,SEC)显示更多样品达到目标寡聚态;其中一例单体获得晶体结构(PDB 8CYK),与目标骨架高度一致(论文报告约 2.35 Å Cα RMSD / 130 残基量级)。
- 环状同源寡聚体:多例环状寡聚体经晶体学或 cryo-EM 验证,骨架与设计模型接近(详见原文与配套工作)。
- 重复蛋白 DHR82:对 Rosetta 重复单元设计不佳的骨架,使用 tying 约束后经 MPNN 序列挽救,AlphaFold 单序列模型与实验行为改善。
- 同时施加环状与重复对称:Rosetta 序列组可溶性约 40% 且 SEC-MALS 无正确寡聚态;ProteinMPNN 组可溶性约 88%,约 27.7% 确认正确寡聚态,并有负染电镜平均投影与设计模型一致。
- 双组分四面体纳米颗粒:在 27 套骨架上设计 76 条序列,13 条表达后形成约 1 MDa 预期分子量的组装;其中一例晶体结构界面与设计模型接近(论文给出约 1.2 Å 量级骨架偏差描述)。
- 功能蛋白设计(Grb2 SH3 结合骨架):Rosetta 序列未表现出预期结合;在相同骨架上经 ProteinMPNN 赋序列后,生物膜层干涉(Bio-Layer Interferometry,BLI)测得与 Grb2 SH3 结构域的强结合信号,且关键突变可消除信号,支持界面编码的特异性。
段末注释:RMSD(root-mean-square deviation)衡量叠加后坐标均方根偏差;SEC-MALS 联用光散射以估计溶液中表观分子量。
8. 典型使用场景与工程实践建议
| 场景 | ProteinMPNN 能做什么 | 常见搭配与注意点 |
|---|---|---|
| 单体稳定化 / 可溶性优化 | 对给定骨架快速生成大量候选序列 | 与 AlphaFold2/ColabFold 单序列回折做预筛;注意表达宿主密码子优化仍影响产量 |
| 蛋白纳米颗粒与疫苗样颗粒 | 利用对称 tying 在多拷贝组装上统一序列约束 | 仍需实验验证颗粒均一性与免疫原性等下游指标 |
| 结合子与酶活性位点周围重塑 | 固定靶标或催化残基,随机顺序解码保留上下文 | 功能位点几何与过渡态偏好常需 Rosetta 或 RFdiffusion 等共设计流程,而非仅靠序列层 |
| 失败设计挽救 | 在不动骨架前提下替换序列,提高可表达与折叠概率 | 若骨架拓扑本身不可实现,序列层无法“无中生有” |
开源实现与权重可从作者团队维护的代码仓库获取(见下节),社区中常与 RFdiffusion(骨架生成)、AlphaFold-Multimer(复合物验证)等组合使用。
段末注释:RFdiffusion 为扩散式蛋白质骨架生成模型,常与序列设计前后衔接形成“生成骨架 → 设计序列 → 结构验证”流水线。
9. 局限与概念边界
- 不直接优化功能:ProteinMPNN 以几何相容与序列可折叠性为主目标;催化、结合亲和力、特异性、免疫原性等需额外筛选或联合其它损失/模型。
- 输入骨架质量敏感但有噪声训练缓解:对低置信度或错误拓扑,任何逆向折叠方法都可能产生“看似合理”的序列;需结合实验或更外层生成模型修正拓扑。
- 指标解释:高序列恢复率不等于设计更“好”,天然序列未必最大化单链可折叠性;论文亦展示 MPNN 序列在 AlphaFold 单序列设置下有时比天然序列更“易预测”回原生骨架。
10. 小结
ProteinMPNN 通过 (1) 丰富主链几何特征 + 边更新 MPNN 编码器、(2) 随机解码顺序带来的任务灵活性、(3) 多链与对称 tying、以及 (4) 面向预测骨架的训练噪声与采样温度策略,把蛋白质序列设计推进到「快速、可扩展、实验可转化」的工程区间。其最大价值在于与当代结构预测与骨架生成工具形成闭环,使研究者能以秒级成本探索大量序列假设,再用实验与更高层模型逐级收敛。
参考文献与链接
- Dauparas J, Anishchenko I, Bennett N, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49–56. doi:10.1126/science.add2187 PMID:36108050 PMC:PMC9997061
- 代码与资源(华盛顿大学 Baker 实验室维护的公开实现,以仓库说明为准):https://github.com/dauparas/ProteinMPNN