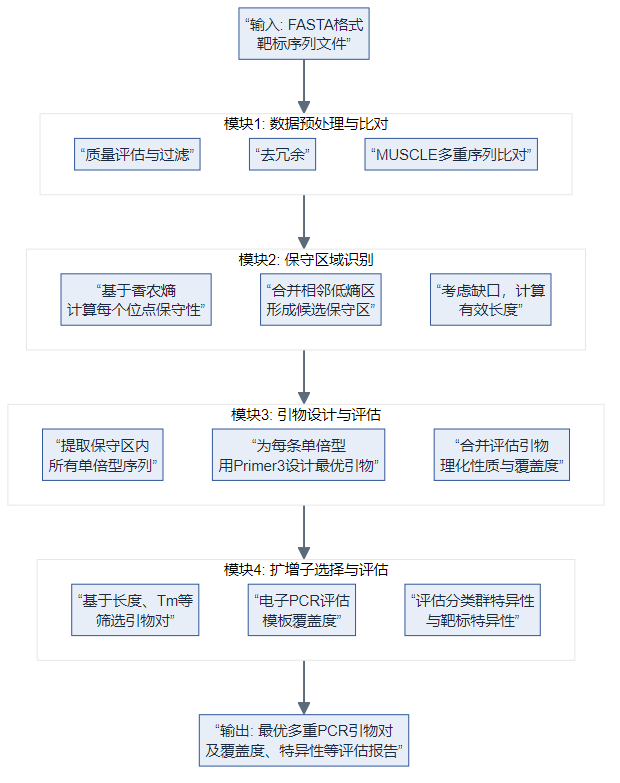
PMPrimer 是 Python 实现的自动化流程:对多样 FASTA 模板做质控与 MUSCLE 比对,用 香农熵 找保守区并容忍 gap,在保守区内按单倍型调用 Primer3 设计简并引物,再评估覆盖度、分类群特异性并可用 BLAST 评靶标特异性(Sci Rep 2023)。
文献与官方资源
| 类型 | 说明 |
|---|---|
| 论文 | Yang L, Lin Q, Xie J, et al. A tool to automatically design multiplex PCR primer pairs for specific targets using diverse templates. Sci Rep 13, 13825 (2023). DOI 10.1038/s41598-023-43825-0 |
| PDF / 补充 | 全文 PDF · MOESM |
| 代码 | https://github.com/AGIScuipeng/PMPrimer |
| 许可证 | 以仓库 LICENSE 为准 |
应用场景
- 多序列模板下的多重 PCR 简并引物设计:如跨物种/跨株系同源区、16S/hsp65/tuf 等数据集(论文示例:葡萄球菌 tuf、分枝杆菌科 hsp65、古菌 16S 等)。
- 需要量化覆盖与分类群区分时:输出 Coverage、Taxon specificity 等列,便于 panel 筛选。
- 不适合:仅单条模板、且无需 MSA/熵筛选的极简场景(用 Primer3 即可);或对实时交互 Web有强依赖时(PMPrimer 以命令行/脚本为主,见仓库)。
使用帮助
部署形态
| 方式 | 说明 |
|---|---|
| 本地 | 克隆 GitHub,按 README 安装 Python 依赖及 MUSCLE5、Primer3、BLAST+(若启用特异性)等;Linux/服务器常见。 |
| 线上 | 无官方统一 SaaS;以仓库说明为准。 |
输入与输出
- 输入
- 主输入:目标序列 FASTA(
seqs.fasta等); - 可选/行为由参数控制:是否过滤过短/过长/重复(
-p notlen、-p notsameseq等关闭默认过滤)。
- 主输入:目标序列 FASTA(
- 中间与输出文件(名称以运行时间与 README 为准)
seqs.filt.fasta:过滤后用于设计的序列;seqs.mc.fasta:启用 MUSCLE 时的多重比对;seqs.matrix.csv/tmp.png:可选距离矩阵与热图(matrix等参数);*_recommand_region_primer.json/.tsv:终表(引物对与评估列)。
资源与环境
- 算力:序列条数多、MSA 与 BLAST 数据库大时,CPU/内存占用显著;论文讨论中提及大内存需求场景,请以实测为准。
- 磁盘:BLAST 库、中间 FASTA/比对文件可能较大。
- 第三方:MUSCLE5、Primer3、BLAST 需单独安装并在 PATH 或配置中可达(见 GitHub)。
功能与算法原理
模块 1:预处理与比对
- 统计过滤:按长度分布等剔除异常与冗余序列(可关)。
- MUSCLE:多重比对,为逐列熵与保守区提供对齐坐标(论文 Methods)。
模块 2:保守区(香农熵 + gap)
- 对对齐后每一列计算 香农熵 (S=-\sum p_i\log_2 p_i),gap 作为独立符号纳入(论文强调)。
- 用主要等位基因频率等阈值将「多保守算保守」落到数值上,合并相邻低熵区段;再扣减 gap 得到 effective length,低于最小长度则丢弃该区。
列熵(公式):设比对后第 (j) 列上符号 (b)(含 A/C/G/T/-)的频率为 (p_{jb}),则
$$
S_j = -\sum_{b \in \mathcal{A}} p_{jb},\log_2 p_{jb},\quad \sum_b p_{jb}=1
$$
(S_j) 越小表示该列越保守。论文将 gap 计入 (\mathcal{A}),避免「全 gap 列」被误判为高保守。
区段合并(示意):将满足 (S_j \le S_{\max}) 且主等位频率 (\ge f_{\min}) 的连续列合并为候选区 ([j_1,j_2]),再计算有效长度(概念上扣除 gap 占比):
$$
L_{\mathrm{eff}} \approx \sum_{j=j_1}^{j_2} \bigl(1 - p_{j,-}\bigr)
$$
其中 (p_{j,-}) 为第 (j) 列 gap 频率;若 (L_{\mathrm{eff}} < L_{\min}) 则丢弃该区(与程序实现阈值以论文/GitHub 为准)。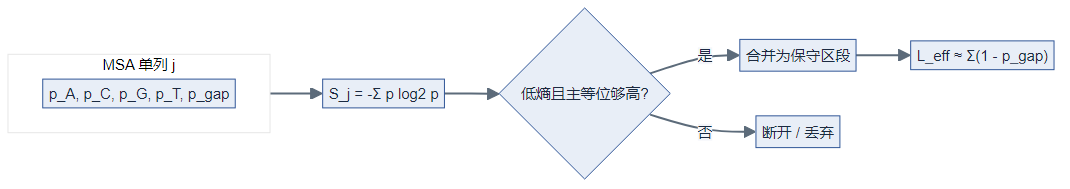
1 | flowchart LR |
模块 3:单倍型 + Primer3
- 不用单条共识序列代替全体模板,而在保守区内抽取真实单倍型路径,对每条单倍型调 Primer3 设计候选,保留稀有等位信息(论文 Fig.1 流程)。
组合规模(示意):若区段内第 (t) 条序列在引物位点上的合法路径视为一条单倍型,对单倍型 (h) 调用 Primer3 得候选集 (\mathcal{P}_h),再对 (\bigcup_h \mathcal{P}_h) 做覆盖与简并合并;简并度与 IUPAC 码由多序列在该窗口上的并集决定。
模块 4:扩增子选择与评估
- 按产物长度、单倍型数、Tm 差等筛多重引物对;电子 PCR 与 BLAST 评估覆盖度与特异性(论文)。
配图(文献原图页面)
| 图号 | 内容(据图题) | 官方页面 |
|---|---|---|
| Figure 1 | PMPrimer 四模块总流程 | Nature — Figure 1 |
| 以下为本地示意图(与论文图互补,非期刊原图;已用统一科研配色由 Mermaid 导出 PNG): | ||
 |
1 | flowchart TD |
输出列(TSV/JSON 摘要)
| 列名 | 含义 |
|---|---|
| Amplicon | 扩增子/区域标识 |
| Coverage | 模板覆盖度 |
| Taxon specificity | 分类群特异性 |
| Effective length | 有效长度(含 gap 处理) |
| Forward/Reverse degenerate primer | 简并引物 |
| Forward/Reverse haplotype primers | 单倍型展开列表 |
| Forward/Reverse primer info | Tm、发夹/二聚体相关度量与覆盖度等(逗号分隔数值) |
局限性
- 依赖链长:MSA、BLAST、大批模板时时间与内存压力大,需分批次或降采样。
- 比对质量:MUSCLE 结果直接影响熵与保守区;错误比对会误导引物区。
- 实验验证:in silico 覆盖度/特异性不能替代湿实验(论文仍强调数据集验证)。
- 维护节奏:以 GitHub issue/提交记录为准;生产环境应固定依赖版本。
参考资料
- Yang L, Lin Q, Xie J, et al. Sci Rep 2023;13:13825. https://doi.org/10.1038/s41598-023-43825-0
- PMPrimer GitHub
- 本站:
引物设计-02.概念-香农熵.md、引物设计-03.软件-Primer3.md