前置阅读:图神经网络:原理、算法与分子建模入门(图、消息传递、PyG 字段);需要三维几何与对称性时再读 SE(3)-等变图神经网络。PDB 文件字段含义可对照 PDB 格式说明。
本文只谈数据层:如何把「一个蛋白结构文件」变成 GNN 能吃的 Data(x, edge_index, pos, …),并对途中每一类 Python 数据结构建立整体印象。
段末注释:PyTorch Geometric(PyG) 是 PyTorch 上的图深度学习扩展库;BioPython 是常用的生物序列/结构解析 Python 包;mmCIF 为 PDB 的现代化文本格式。
1. 端到端数据流(先建立地图)
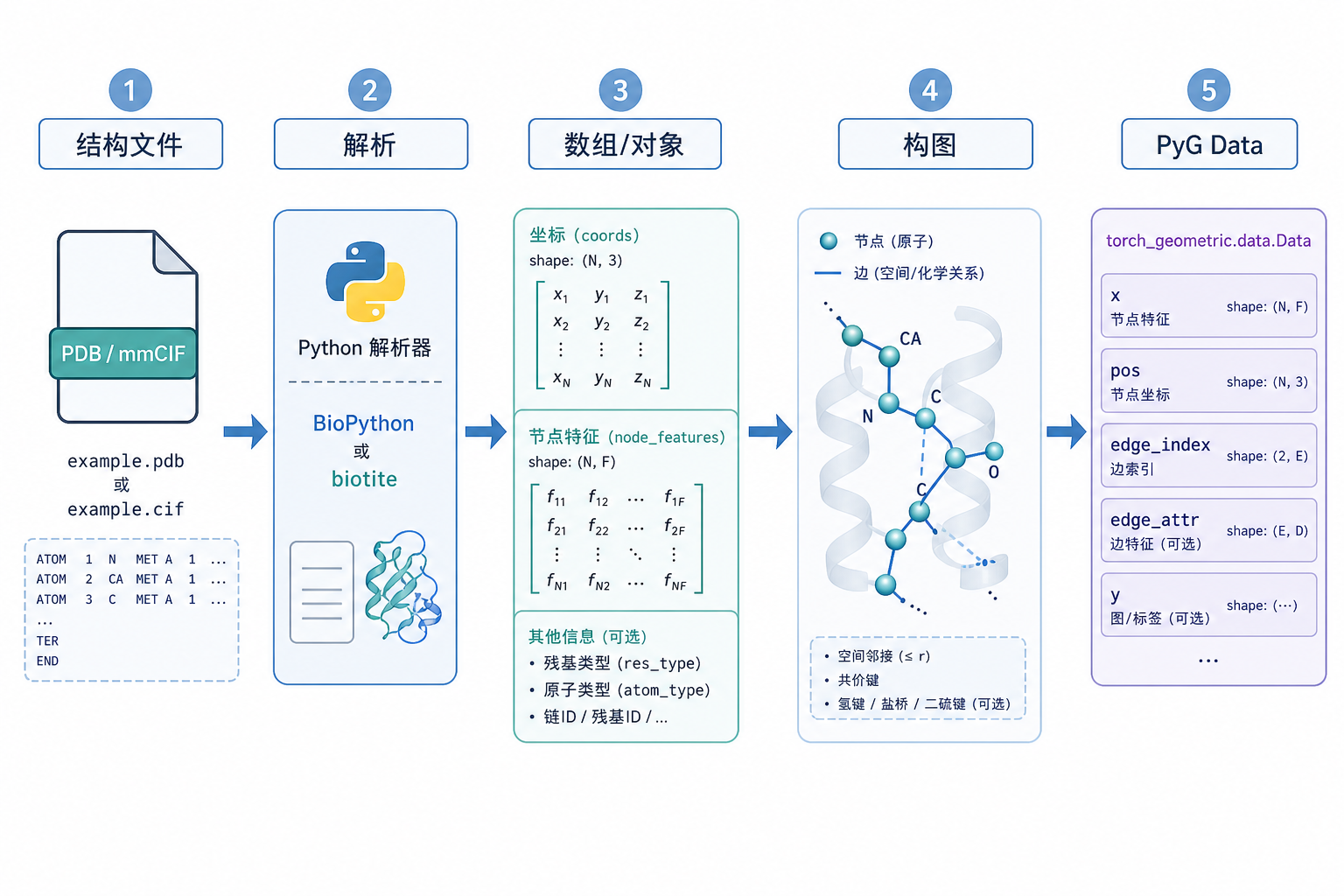
图 1(科普示意):实践中的主路径可概括为五步。
| 步骤 | 输入 | 输出 | 典型 Python 类型 |
|---|---|---|---|
| ① 读文件 | .pdb / .cif |
文本记录 | str、路径 Path |
| ② 解析 | ATOM/HETATM 行 | 层次化结构对象 | BioPython Structure |
| ③ 抽表 | 选定原子/残基 | 坐标与特征表 | numpy.ndarray、torch.Tensor |
| ④ 构图 | 坐标 + 规则 | 邻接关系 | edge_index [2, E] |
| ⑤ 封装 | 张量集合 | 单样本图 | torch_geometric.data.Data |
结构编码在 GNN 语境下指:用图 $G=(V,E)$ 把三维构象离散化,并把每个节点 $i$ 编成特征向量 $\mathbf{x}i$(可选边特征 $\mathbf{e}{ij}$、坐标 $\mathbf{p}_i$),供消息传递学习 $\mathbf{h}_i$ 乃至图级 $\mathbf{h}_G$。
2. 结构文件里有什么
实验或预测结构常见 PDB(Protein Data Bank 文本格式)与 mmCIF(macromolecular Crystallographic Information File)。对 GNN 最重要的是:
- 谁:残基名、链 ID、残基序号、原子名(
CA= $C_\alpha$) - 在哪:$x,y,z$(Å)
- 可选置信度:预测结构常把 pLDDT(predicted Local Distance Difference Test)写在 B-factor 列
一条 ATOM 行(概念分区,列宽以 wwPDB 文档 为准)包含:原子坐标、占有率、温度因子 B 等。GNN 构图时通常不把整行原样喂进网络,而是抽取成数值张量。
3. Python 中的结构表示:三层结构
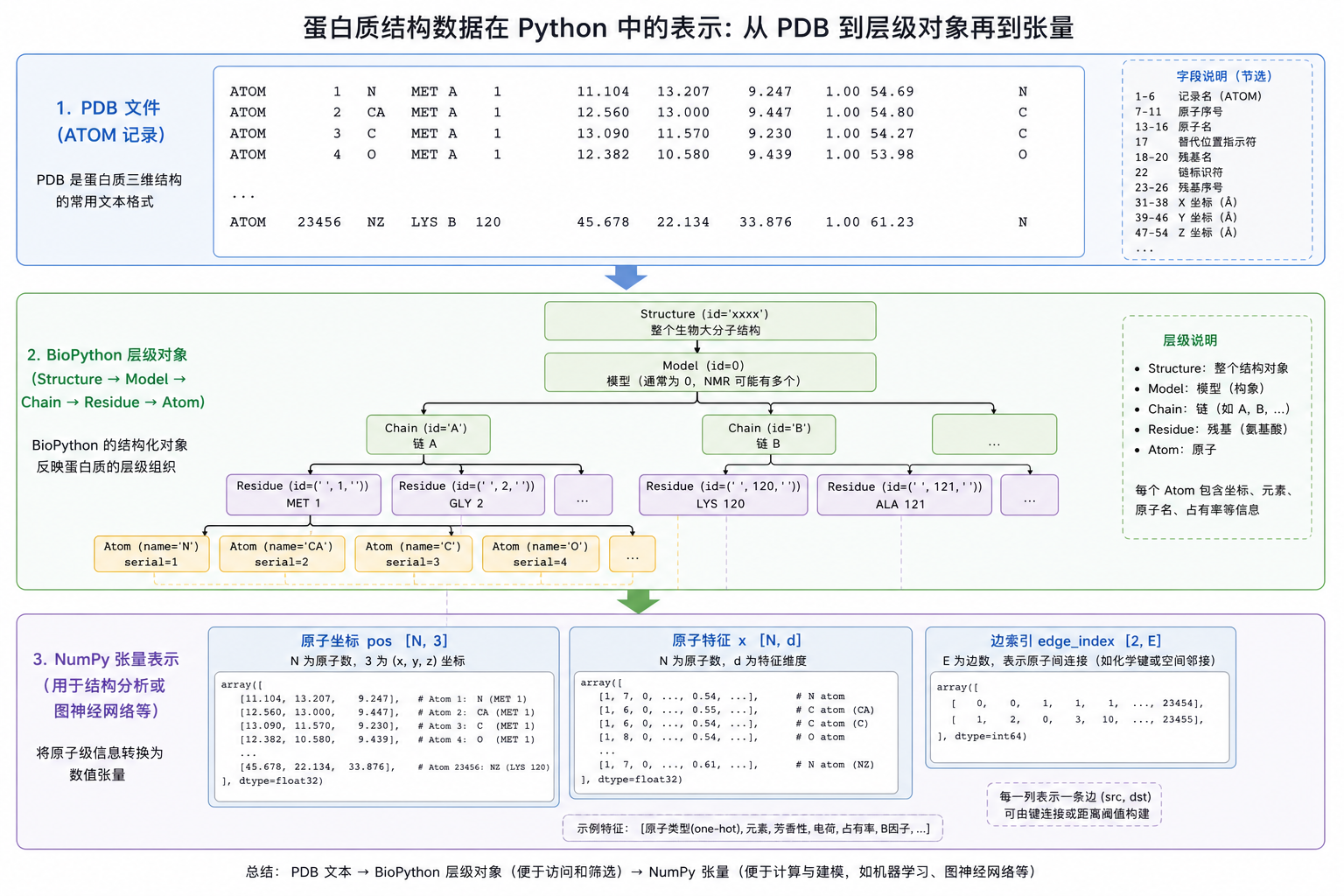
图 2(科普示意):同一份结构在内存里会同时存在「对象树」与「扁平张量」两种视图,后者才直接对接 GNN。
3.1 层次对象(BioPython)
BioPython 的 PDB 解析器(Bio.PDB.PDBParser)把文件读成:
1 | Structure(id) # 一个文件常对应一个 Structure(或 NMR 多模型) |
- 适合:按链筛选、跳过 HETATM、访问
residue.get_resname()等语义操作 - 不适合:直接做批量矩阵运算(需再导出为数组)
3.2 扁平张量(NumPy / PyTorch)
GNN 实际使用的是「表格式」数据:
| 张量 | 形状 | 含义 |
|---|---|---|
pos |
[N, 3] |
节点坐标,常取 $C_\alpha$ |
x |
[N, F] |
节点特征(氨基酸类型 one-hot、B-factor、pLDDT 等) |
edge_index |
[2, E] |
源节点、目标节点索引(COO 格式) |
edge_attr |
[E, D_e] |
边长、序列距离、接触类型等 |
seq_pos |
[N] |
序列位置 0…$L{-}1$(可选) |
3.3 其它常用库(选型)
| 库 | 特点 | 典型用途 |
|---|---|---|
| BioPython | 对象树清晰、教程多 | PDB 解析、快速原型 |
| biotite | 数组优先、AtomArray |
高性能筛选、几何运算 |
| MDAnalysis | 轨迹、大规模动力学 | MD 轨迹 → 接触图 |
| ASE | 更偏材料/通用原子 | 与化学模拟衔接 |
下文示例以 BioPython + PyTorch + PyG 为主,换 biotite 时只需把「对象树 → pos/x」一步改成 AtomArray 索引。
4. 步骤 ①②:解析 PDB 并提取 $C_\alpha$
1 | from pathlib import Path |
此时得到的是序列对齐的残基表:节点数 $N = L$(有 $C_\alpha$ 的残基数),尚未定义「边」。
段末注释:$C_\alpha$ 为每个氨基酸骨架上的 $\alpha$ 碳,蛋白残基图的常用节点;全原子图以所有重原子为节点,$N$ 约为 $4\text{–}10 \times L$。
5. 步骤 ③:节点特征 x 怎么编
常见编码方式(可组合拼接):
| 特征块 | 维度 | 说明 |
|---|---|---|
| 氨基酸类型 | 20 或 21 | one_hot(aa) 或嵌入表 nn.Embedding |
| 序列位置 | 1 或 $d_{pe}$ | 归一化残基索引,或正弦位置编码 |
| 置信度 | 1 | B-factor / pLDDT,归一化到 $[0,1]$ |
| 二级结构 | 3 | DSSP 类别 one-hot(需额外计算) |
| MSA 统计 | 变长 | 保守性、共进化(需多序列比对) |
1 | import torch.nn.functional as F |
pos 单独存放,不强行并入 x:几何 GNN / SE(3)-等变 模型需要显式坐标做距离、方向消息(见续篇)。
6. 步骤 ④:从坐标到 edge_index
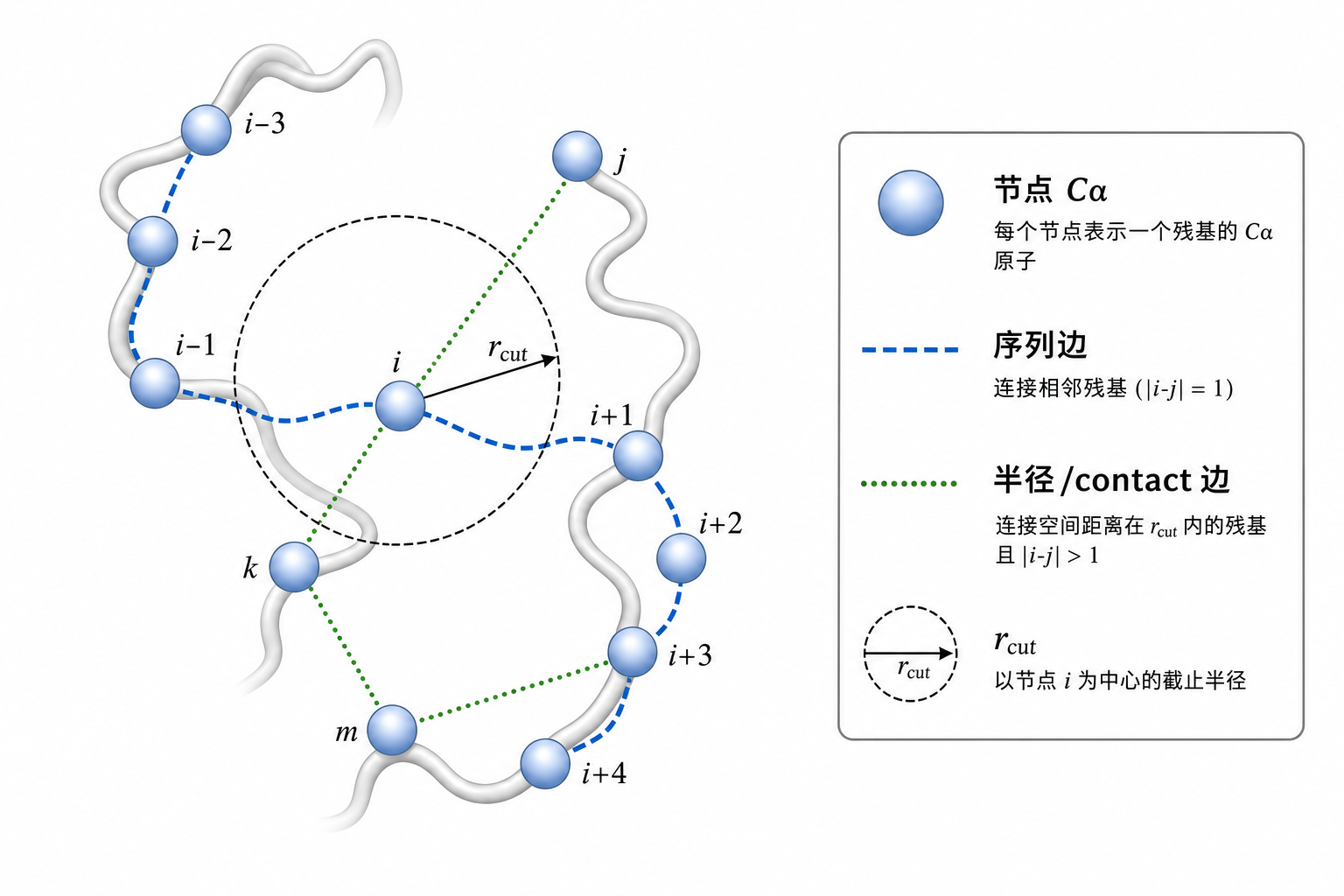
图 3(科普示意):蛋白残基图常同时使用序列邻接与空间邻接。
6.1 序列边(化学拓扑先验)
相邻残基 $i$ 与 $i{+}1$ 连无向边(实现上双向各一条):
1 | def sequential_edges(num_nodes: int) -> torch.Tensor: |
6.2 半径图(空间接触)
当 $r_{ij} = |\mathbf{p}_i - \mathbf{p}_j|2 < r{\mathrm{cut}}$ 时连边(Å 单位,常用 $8\text{–}12\ \mathrm{\AA}$ 残基级、全原子更短):
1 | from torch_geometric.nn import radius_graph |
也可用 torch_geometric.nn.knn_graph(pos, k=30) 控制每节点邻居数上界。
6.3 合并边并去重
1 | def merge_edges(*edge_indices: torch.Tensor) -> torch.Tensor: |
6.4 边特征 edge_attr(可选)
1 | def edge_length_attr(pos: torch.Tensor, edge_index: torch.Tensor) -> torch.Tensor: |
序列边可附加 $|i-j|$(序列距离);空间边以 $r_{ij}$ 为主。带方向的模型还会用单位向量 $\hat{\mathbf{r}}_{ij}$(等变 GNN)。
7. 步骤 ⑤:组装 torch_geometric.data.Data
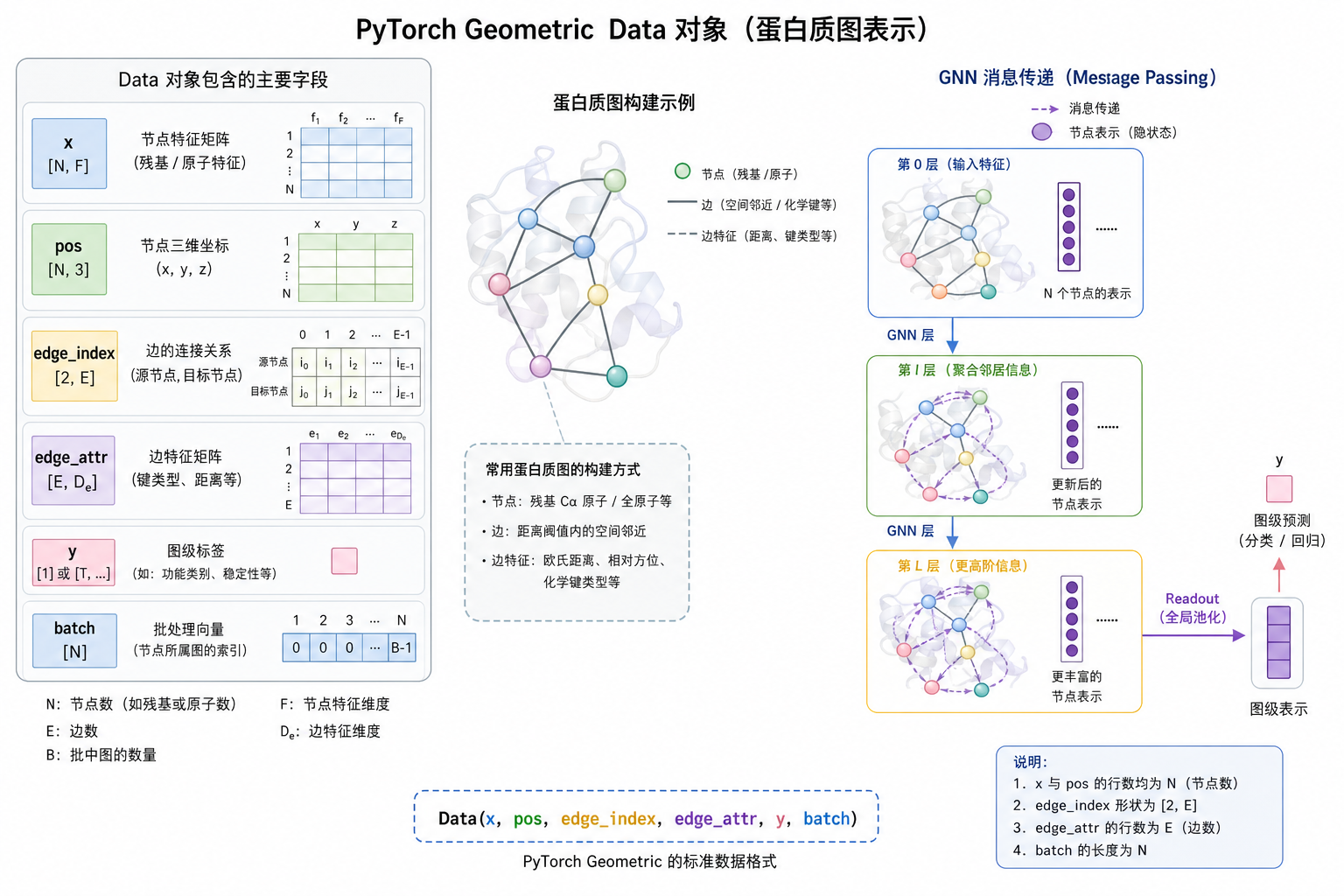
图 4(科普示意):Data 是单张图的「容器」;forward 时由 GNNConv 读 x 与 edge_index。
1 | from torch_geometric.data import Data |
7.1 Data 与数学对象对照
| PyG 属性 | 数学/用途 |
|---|---|
data.x |
节点特征矩阵 $\mathbf{X}$,$\mathbf{h}_i^{(0)}=\mathbf{x}_i$ |
data.pos |
$\mathbf{p}_i \in \mathbb{R}^3$,不必然进入首层 MLP |
data.edge_index |
邻接的 COO 列表,定义 $\mathcal{N}(i)$ |
data.edge_attr |
$\mathbf{e}_{ij}$,进入 $\phi_m$ |
data.y |
图级标签 $y_G$ |
data.batch |
批训练时标记节点属于哪张图 |
8. 批训练:多蛋白如何拼成一批
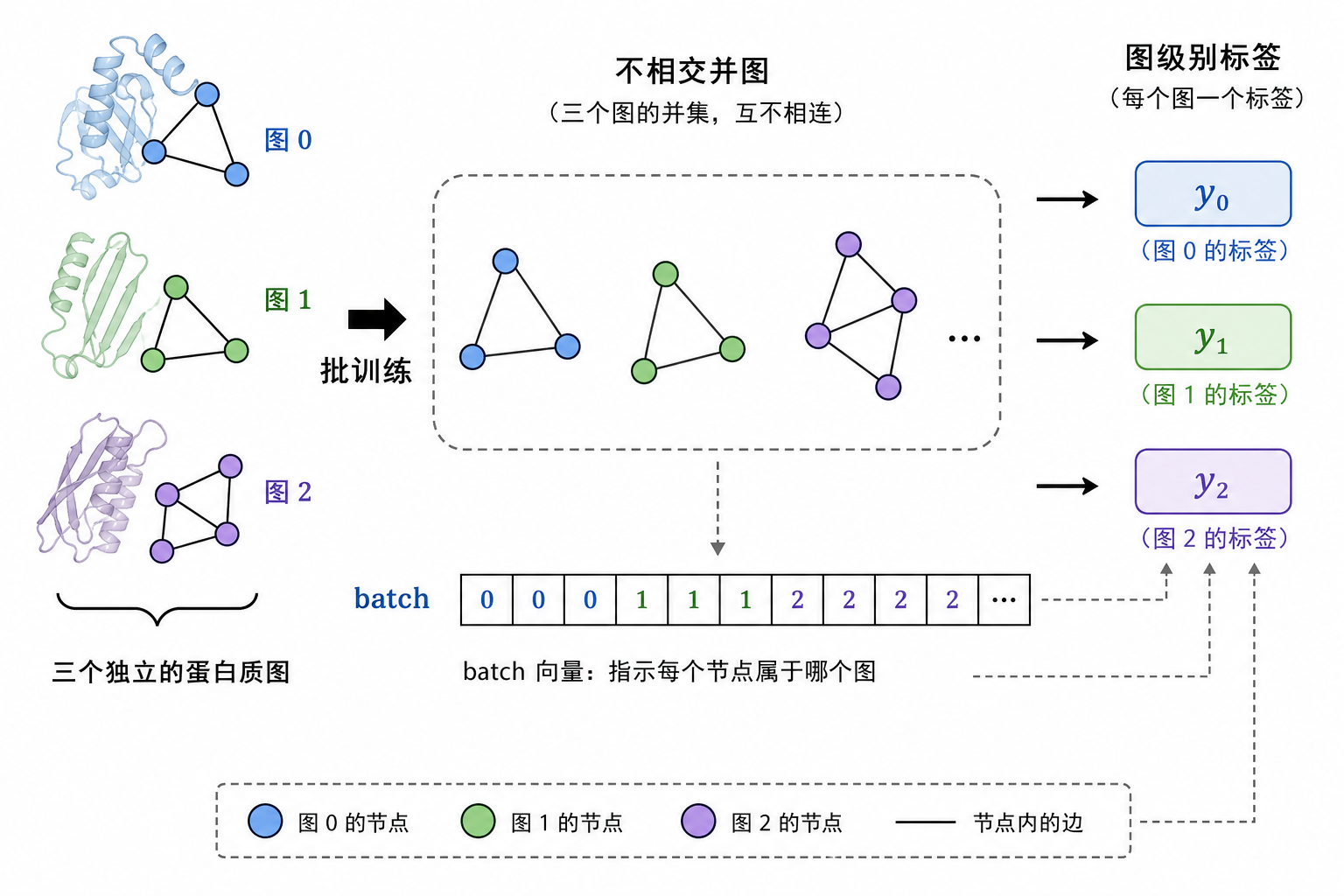
图 5(科普示意):PyG 的 DataLoader 会把多张图并成一个大图(节点不相连),用 batch 向量区分。
1 | from torch_geometric.data import DataLoader |
Dataset 模式:把 structure_to_pyg_data 放进 torch_geometric.data.Dataset 子类,在 get(idx) 里惰性解析 PDB,避免一次性载入万级结构。
9. 数据结构总览(速查表)
| 阶段 | 你手里的东西 | 关键索引语义 |
|---|---|---|
| 文件 | PDB/mmCIF 文本 | 残基序号、链、原子名 |
| BioPython | Residue / Atom |
res["CA"].coord |
| NumPy | pos[i], aa[i] |
$i$ 与序列顺序一致 |
| 图 | edge_index[:, e] |
edge_index[0,e] -> edge_index[1,e] |
| PyG | Data |
data.num_nodes == data.x.size(0) |
| Loader | Batch |
data.batch[i] = 第 $i$ 个节点所属图的 batch 内编号 |
索引一致性:edge_index 中的值必须在 $[0, N-1]$ 内;合并多源边后建议 torch.unique;过滤低 pLDDT 残基后要同步删 pos/x 的行并重映射边。
10. 蛋白结构编码的常见建模选择
| 决策 | 选项 A | 选项 B | 影响 |
|---|---|---|---|
| 节点 | $C_\alpha$ 残基 | 全原子 | $N$ 规模、边定义 |
| 边 | 仅序列 | 序列 + 半径图 | 是否捕获远程接触 |
| 坐标 | 只作构图 | pos 进等变层 |
是否需 $\mathrm{SE}(3)$ 模型 |
| 标签 | 图级(稳定性) | 节点级(RSA) | y 形状与损失 |
| 划分 | 随机 | 按 PDB ID / 簇 | 是否泄漏同源蛋白 |
编码的最终产物是:在保留几何与序列先验的图上,每个残基有一个初始嵌入 $\mathbf{x}_i$;后续 GNN 层通过消息传递得到 $\mathbf{h}_i$、$\mathbf{h}_G$,用于活性预测、界面识别、突变效应等下游任务。
11. 最小可运行依赖
1 | pip install biopython numpy torch torch-geometric |
1 | 项目目录示例 |
12. 与系列文章的衔接
| 主题 | 本文 | 其它篇 |
|---|---|---|
| 图与 MPNN | edge_index、x |
GNN 入门 |
| 旋转等变 | pos 进网络 |
SE(3)-等变 GNN |
| PDB 列含义 | ATOM、B-factor | fileformat-pdb.md |
| 抗体域级结构 | 选链、Fab/Fc | 抗体结构 |
13. 小结
用 Python 做蛋白结构的 GNN 编码,本质是 「结构文件 → 残基表 → 图张量 → PyG Data」。BioPython(或 biotite)负责语义解析,NumPy/PyTorch 负责数值表,edge_index 负责关系,PyG 负责与 GNN 层对接。记住三张图:对象树(便于筛链筛残基)、坐标表 pos(几何与构图)、特征表 x(化学与统计),再记住两种边:序列与空间,就能把文献里的蛋白 GNN 数据管线读懂、写通。
参考文献与延伸阅读
- BioPython Tutorial — Structure section
- PyTorch Geometric — Creating Your Own Datasets
- Hamelryck & Manderick, PDB parser and structure class (BioPython PDB).
- 本系列:图神经网络入门、SE(3)-等变 GNN