DNA是遗传物质,其重要性不言而喻,针对DNA、RNA和蛋白的研究也层出不穷。之前有很多基于Transformer的DNA模型,受限于起本身上下文长度的限制,经常采用寡居核苷酸而不是单碱基所谓模型训练/学习的基本单位。而本问开发的Evo模型,是一个包含7-billion-参数 的模型,训练以用来在全基因尺度上进行DNA序列的生成。该模型的基础架构是基于 StripedHyena architecture。 该模型结合了注意力模型和针对数据的卷积核,来高效的处理长序列数据。Evo的训练集是由 300 bilion 原核全基因组的核酸组成的全基因组数据集上进行训练的,并对输入数据进行单碱基层面的分词。并且发现预测表现伴随着数据规模的增大而获益。
Materials and method
Evo 基于 StripedHyena。
- Evo包含32个block,模型宽度为4096维。
- 每个块包含一个序列混合层,负责沿序列维度处理信息,以及一个通道混合层,侧重于沿模型宽度维度处理信息。
- 在序列混合层中,Evo使用29个鬣狗层,并在其中等间隔的穿插交错着3个旋转自关注层交错。我们使用参考文献中描述的模态规范形式参数化鬣狗算子中的卷积。
- 对于通道混合层,Evo使用门控线性单元。
- 使用均方根归一化每个输入层。
Hyena layers/鬣狗层
鬣狗 是一个序列混合器,由短卷积、长卷积和一个数据控制门 构成,结构如下: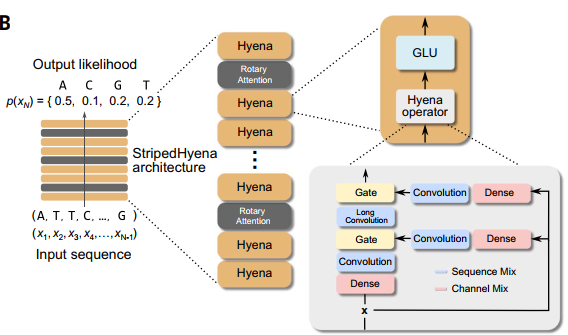
Self-attention layers / 自注意力层
自注意力将输出序列构造为输入元素的加权组合,其中的权重由输入决定。
Positional embeddings / 位置嵌入
自注意算子对输入序列中输入嵌入的不同位置没有任何概念。
出于这个原因,它通常会辅以位置编码机制。StripedHyena的注意层使用旋转位置嵌入机制(RoPE)来模拟相对位置信息
在我们的第二个预训练阶段,我们应用线性位置插值来扩展在第一个预训练阶段(8k序列长度)应用的旋转位置嵌入[Extending context window of large language models via positional interpolation]。当应用于比最初训练时更长的序列时,插值使模型能够继续利用其学习到的表示。我们还测试了其他位置插值方法,但发现它们在数据上的表现略差于线性插值
Tokenization / 分词器
Evo使用Python中实现的UTF-8编码,以单核苷酸分辨率对DNA序列进行标记。在预训练期间,Evo使用了一个有效的词汇表,包含四个标记,每个碱基一个,总共有512个字符,这允许在后续的下游任务中扩展词汇表。我们使用额外的字符来启用在使用微调模型生成过程中使用特殊令牌的提示
OpenGenome datasets
(i) the Genome Taxonomy Database (GTDB) 的细菌和古细菌数据;
(ii) theIMG/VR v4 database (36) 的筛选后的原核病毒;
(iii) IMG/PR database (37) 的质粒序列数据。
所有的数据均表示直接拿来使用的, 数据均进行过相应的筛选和过滤。因为这部分实际用途不大,有需求查看原文,此处略过。
Training procedure
整个Evo的训练过程分两个阶段,第一阶段是使用80k token的上下文,第二阶段上下文提升到131B token(分阶段是为了提高训练的效率)。累计训练了4周 * 2线程 (we trained Evo in stage 1 on 64 NVIDIA H100 GPUs for 2 weeks and on 128 NVIDIA A100 GPUs in stage 2 for an additional 2 weeks.)
Dataloading
序列打包生成训练数据,每个特定上下文的序列是从完整的序列数据集中随机采样的。
一些DNA序列比上下文长度短,这时把多个DNA的序列连接起来,直至长度满足预期(8k 或 131k)。如果DNA序列比上下文长度长,就会截取基因组的子序列。
Hyperparameter tuning and direct model comparisons
测试了 7B Transformer++,Hyena 和 StripedHyena, StripedHyena具有整体最低的困惑度和损失函数。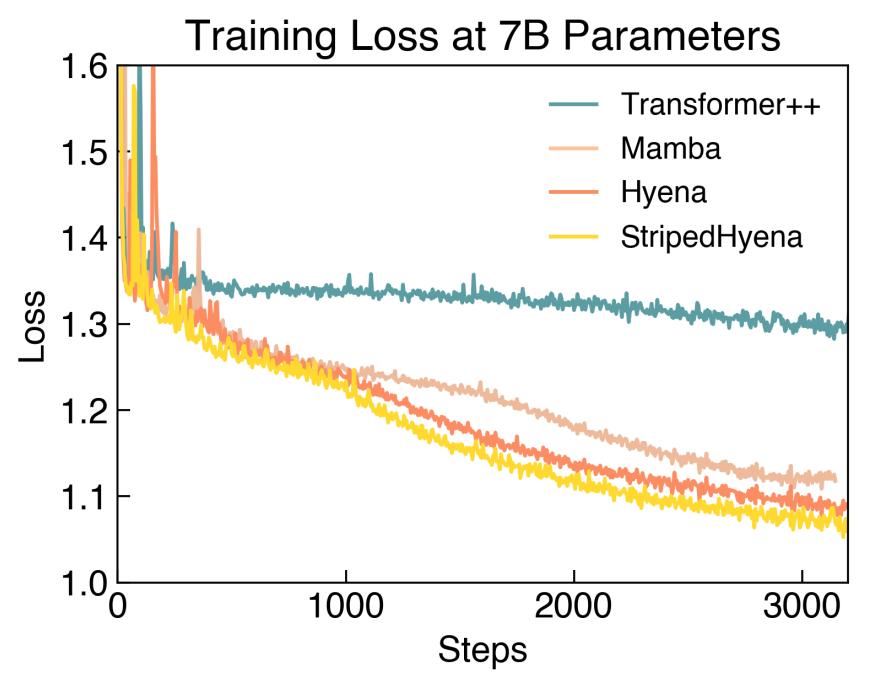
Scaling laws
我们通过计算最优协议比较不同类型的架构,旨在评估计算最优前沿的结果。计算优化分析研究给定计算预算(通常以浮点运算(FLOPs)表示)的预训练运行的最佳性能,并通过将部分计算预算优化分配给模型大小和数据集大小来实现。体系结构类型在计算效率以及如何分配计算预算方面有所不同。我们首先通过网格搜索调优Transformer++的学习率和批处理大小等超参数,然后对所有架构使用相同的值,除了观察到数值不稳定的设置。为了解决不稳定性问题,我们逐渐降低学习率并重复实验直到收敛。在所有实验中,我们训练了上下文长度为8192个令牌的模型。对于由总FLOP计数定义的每个计算预算,我们改变了模型大小(600万到10亿个参数)和训练的令牌数量。为了测量模型性能,我们使用困惑度度量,它表明自回归模型在预测序列的下一个标记方面的表现如何,并且与下游任务的性能高度相关。perplexity值越低,性能越好。
Scaling laws procedure
1) 确定预算. We use $8 × 10^{18}, 2 × 10^{19}, 4 × 10^{19}, and 8 × 10^{19}$ FLOPs (floating point operations)
2) 计算要使用的模型架构处理输入数据所需要的 FLOPs
3) 确定每个计算预算下模型的最优分配:
a) 模型的大小,选择比较宽的范围,然后计算每个模型大小计算需要处理的相应令牌数量以达到计算预算。根据表S3选择其他超参数。我们通常观察到模型拓扑结构(深度,宽度)的微小变化只会最小程度地影响perplexity。
b) 训练每种大小的模型并记录其表现(例如,在困惑度方面)。
c) 确定最优计算分配:根据之前的分析,我们拟合一个二阶多项式作为(log)模型大小与perplexity的函数,并提取得到的计算最优点作为其最小值。计算最优点在给定的计算预算下确定模型大小和训练令牌的最佳分配。
在得出计算最优缩放率之后,我们比较了体系结构并计算了令牌和模型大小的最优分配(图S5)。在图S3中,我们还显示了按架构计算次优模型尺寸的比率。我们量化了计算预算分配到模型或数据集大小的次优分配(例如,为更多令牌训练较小的模型)对困惑缩放的影响。我们估计每个计算预算的计算最优模型大小,然后将其减少一个百分比(偏移量)。相应的perplexity通过IsoFLOP曲线得到(图1F)。与Hyena和StripedHyena相比,Transformer++的复杂度缩放在计算最优边界之外迅速退化。表S3提供了为我们的缩放律分析而训练的模型的架构细节
Transformer++
我们使用具有旋转位置嵌入、均方根层归一化的预处理和SwiGLU作为激活函数的现代 decoder-only Transformer架构。SwiGLU的内部宽度是模型宽度的4/3。我们对分组查询注意(GQA)进行了实验(117),发现最终损失的差异很小,这表明该技术可能适合DNA序列建模,以进一步减少推理过程中的内存占用。所有使用Transformer++的缩放结果都不使用GQA。
Hyena
Hyena基线的设计采用了与Transformer++模型相同的体系结构改进。我们将所有多头自注意层替换为鬣狗层,并对状态维为8的长卷积使用模态规范参数化
Protein function prediction
我们使用DMS数据集来测试蛋白质和核苷酸语言模型预测蛋白质功能突变效应的能力。在所有情况下,我们都使用了原始研究作者报告的核苷酸序列。我们的分析仅限于原核和人类蛋白质,其中值得注意的是Evo训练数据集仅包含原核蛋白质序列。
为了编译来自原核生物DMS研究的核苷酸信息,我们使用了ProteinGym基准中列出的所有“原核生物”数据集,我们也可以找到原始研究作者报告的核苷酸水平信息。这导致了9项研究:Firnberg等人(118)的黑酰胺酶DMS, Jacquier等人(119)的黑酰胺酶DMS, CcdB DMS(120),多蛋白热稳定性数据集(121),IF-1 DMS (122), Rnc DMS (123), HaeIII DMS (124), VIM-2 DMS(125)和APH(3 ‘)II DMS(126)。
为了编译来自人类DMS研究的核苷酸信息,我们将人类基准中使用的数据集的范围缩小到参考文献(45)中用于基准突变效应预测因子的人类数据集。我们还将分析限制在我们也可以找到原始研究作者报告的核苷酸水平信息的研究中。这导致了六项研究:CBS DMS (127), GDI1 DMS (128), PDE3A DMS (129), Kotler等人的P53 DMS (130), Giacomelli等人的P53 DMS(131)和BRCA1 DMS(132)。
我们将Evo(在8k背景下进行预训练)与两个基因组DNA语言模型进行了比较:GenSLM 2.5B,前者使用原核生物基因组序列的密码子词汇表进行训练(15),而Nucleotide Transformer 2B5_multi_species则使用原核和真核生物基因组序列的6 mer核苷酸词汇表进行训练(16)。我们还将Evo与几种在非冗余的蛋白质序列通用语料上训练的蛋白质语言模型进行了比较:CARP 640M(46)、ESM-1v(41)、esm - 2650m、esm - 23b(47)、ProGen2 large和ProGen2 xlarge(48)。对于提供具有多个参数大小的模型的研究,我们选择了可以在不超过GPU内存的情况下使用80 GB NVIDIA H100 GPU对所有基准研究序列进行推理的最大大小。我们还纳入了ESM-2 650M和ProGen2 large,因为这些模型有时在功能预测方面比这些模型的大版本表现得更好(44)。
为了比较核苷酸和蛋白质语言模型,我们使用了原始研究报告的所有独特的核苷酸序列及其相应的适应度值。偶尔,我们观察到报告的核苷酸序列的适应度值与报告的蛋白质序列的适应度值不同;在这种情况下,我们使用报告的核苷酸序列的适应度值,并使用翻译的序列评估蛋白质语言模型。如果由于密码子的使用不同,单个蛋白质序列存在多个核苷酸序列,则在每个唯一核苷酸序列上评估核苷酸语言模型,并在每个唯一核苷酸序列对应的编码序列上评估蛋白质语言模型;这意味着一个蛋白质语言模型可以在一个给定的研究中对相同的蛋白质序列进行多次评估。一些研究报告了涉及终止密码子的突变的适应度值;在这种情况下,我们评估了包含停止密码子的序列的核苷酸语言模型,并将这些例子从蛋白质语言模型基准中排除。我们计算了实验适应度值与序列似然(用于自回归语言模型)或序列伪似然(用于掩码语言模型)之间的Spearman相关性。当使用Evo序列似然对序列进行评分时,我们还将EOS令牌(在预训练数据中用于划分不同序列)前置到完整序列,我们从经验上发现这可以提高零射击性能。我们在零假设下评估了Spearman相关系数的统计显著性,即相关系数来自具有N - 2个自由度的t分布,其中N是我们计算相关性的样本数量。我们使用这个零分布来根据观察到的相关性计算P值。我们使用scipy Python库(https://scipy.org/)来计算这些值。