官方资料
github: ClinCNV
github: pre-process ngs-bits
ClinCNV: multi-sample germline CNV detection in NGS data
流程说明
部署应用
项目代码拉取
1 | CNV 分析项目代码包 |
R环境安装基于conda
创建R基础环境,github说明是基于 3.2.3 版本。但是3.2.3 版本已经不维护。。
中间版本可能存在问题:
- R=3.6.1
“MASS”、”party”、”umap” 编译有问题,无法通过install.packages安装。其中part的依赖包 mvtnorm安装问题
安装问题。未检索到直接解决方案,
经测试相关包安装可以在目前最新的R编译版本(R=4.4.1)中,可以通过install.packages完成全部依赖包的自动安装。
1 | conda create -nClinCNV -c conda-forge r-base=4.4.1 |
R包安装
完成镜像后,进入R环境安装所列的依赖包
1 | # 能直接安装的 |
R包安装确认
测试包安装
1 | # 能直接安装的 |
demo数据运行
1 | 设置本地拉取的ClinCNV项目存储路径 |
可以看到流程读取两个输入文件 .cov 是一个覆盖度的矩阵文件(包含待分析的所有样本). .bed 进行了GC-含量注释的bed文件 (from 0 to 1, should be in 4th column).
bed_file.bed
bed文件时在第四列注释了GC含量,第五列标注基因名称信息(第四列是必须的,第五列是可选的)。bed文件中不应该包含文件头,或文件头应该是用 # 注释的。
| 列号 | 说明 | 备注 |
|---|---|---|
| 1 | 染色体 | 染色体位置,chr前缀 |
| 2 | 起始坐标 | int |
| 3 | 结束坐标 | int |
| 4 | GC含量 | real, from 0 to 1 |
| 5 | genes | character, 逗号分隔,可选内容 |
1 | chr1 12171 12245 0.4595 |
coverages_normal.cov
.cov 文件是包含染色体区域信息(前三列),每个样本的对应区域的覆盖度(每个样本一列,建议是区域的平均覆盖度)矩阵文件。首行是文件的头文件。
| 列号 | 说明 | 备注 |
|---|---|---|
| 1 | 染色体 | 染色体位置,chr前缀 |
| 2 | 起始坐标 | int |
| 3 | 结束坐标 | int |
| 4 | Sample1 | 样本在对应区段的平均覆盖深度 |
| …. | …. | ….. |
| 3+n | sample-n | 样本在对应区段的平均覆盖深度 |
1 | chr start end Sam1 Sam2 |
文件 *.cov生成
建议使用samtools进行区域覆盖深度的生成,一方面这是 clinCNV 官方文档中提供的一个计算覆盖度的方式,另一方面是samtools在大多数NGS分析过程中都会用到,所以一般不需要我们进行额外的安装。
基于samtools
统计深度的命令格式如下:
1 | samtools bedcov temp.bed pancancer689__DX1790_daiyufen_23S03853625_23B03853625__Cancer.realign.bam |
基于ngs-bits(不建议)
ngs-bits 是 clinCNV 官方文档中提供的一个计算覆盖度的方式,但是安装过程发现 ngs-bits 安装的底层依赖和最新版的 clinCNV 已经存在冲突。所以建议更换掉 ngs-bits 软件。
- 安装
1
conda create -n ngs-bits -c bioconda ngs-bits
- 使用
1
BedCoverage -bam $bamPath -in $bedPath -min_mapq 3 -out $sampleName".cov"
mosdepth
mosdepth 是另外一个可以实现相关功能的软件。
- 安装
1
conda install -c bioconda mosdepth
- 使用
1
mosdepth --by temp.bed output_prefix input.bam
- 结果
运行后会生成多个结果,其中regions.bed.gz,就是可以用ClinCNV进行CNV分析的格式文件(单样本),对每个样本进行处理后,需要对多个样本的结果进行整合。
结果说明
批次统计结果
批次结果有两部分,1. 统计每个样本的平均深度信息,并计算样本的内部噪音;2.对批次内地所有样本,计算样本间的相关性,完成聚类(聚类使用umap默认参数,聚类数目为15,所以所提供的样本需要超过16例样本。
样本聚类情况统计
测试数据载入后,会进行前期的聚类分组,聚类结果见 $fold"/results/clusterization_of_samples.tsv 会输出聚类后所有样本的cluster分类情况(sample_id,cluster_id)
1 | 20240827__***********47 0 |
同时也会生成聚类图$fold"/results/clusteringSolution.png"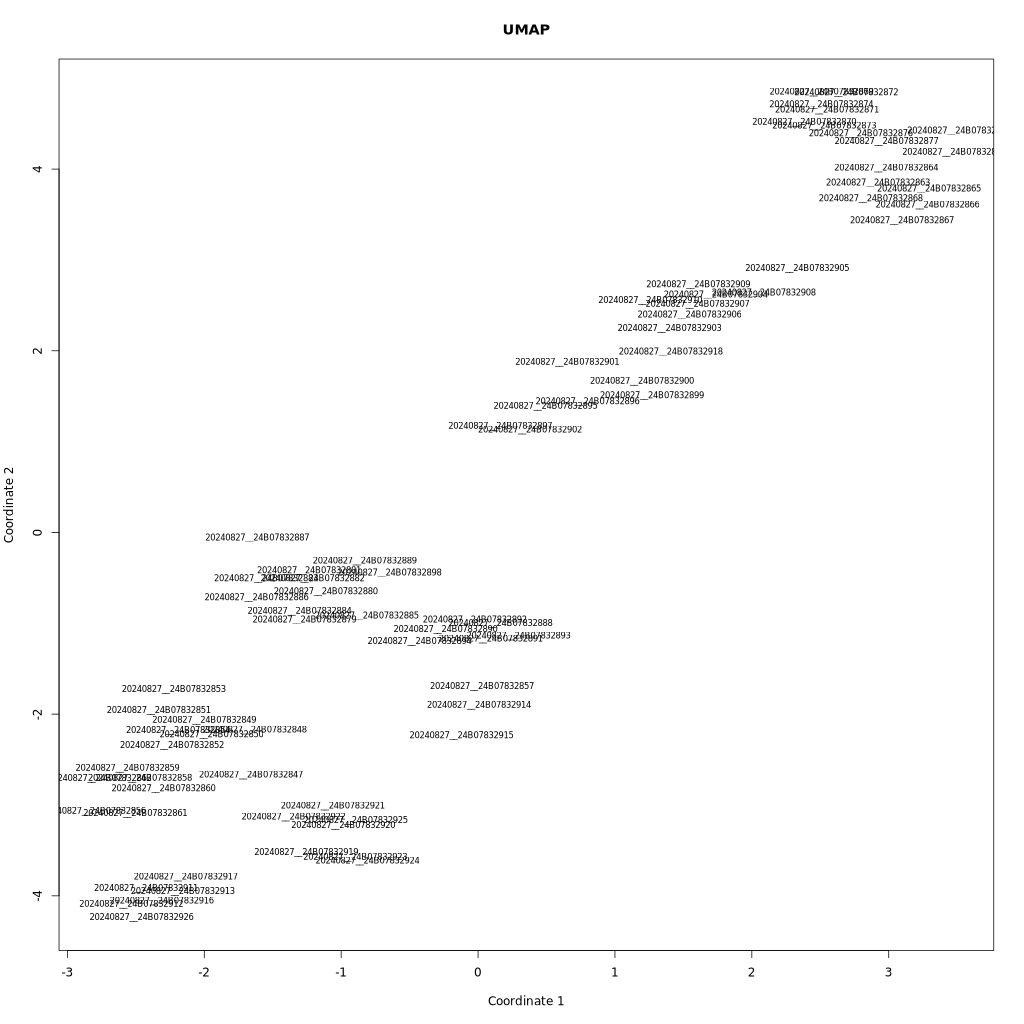
样本覆盖度情况统计
$fold"/results/ontargetNormal.summary.xls" 会统计整批分析样本的平均深度信息以及计算得到的噪音信息(noises <- round(apply(gcNormalisedCov[which(!bedFile[,1] %in% c(“chrX”,”chrY”)),], 2, mad), digits = 2))。
1 | namesOfOutputFile noises avgDepth |
样本CNV检测结果
带 cnvs 标记的,都是样本的CNV检测结果对应的信息,其中 tsv 文件为 ClinCNV 输出的CNV详细信息(涵盖了bin的输入,CNV的长度,覆盖的基因等。
两个 seg 后缀的文件为IGV的tracks文件,
- _cov.seg,提供了实际覆盖率的值(覆盖率值是 GC 归一化和中位数归一化,以常染色体中的拷贝数 2 为中心)
- _cnvs.seg文件中,拷贝数范围只有 [0,1,2,3,4,5,6] ,所有6拷贝以上的CNV,都按6进行输出。
******_cnvs.tsv
最主要的CNV结果,结果由两部分构成,标签标题行和结果表格。
标签标题行示例如下:
1 | ##ANALYSISTYPE=CLINCNV_GERMLINE_SINGLE |
- 前 1 行 是技术性的记录。
- 前 2 行 是软件版本。
- 前 3 行 是分析的时间。
- 第 4 行 显示推断的性别。
- 第 5 行 自从样本中的 CNV 数量变得可接受以来,质量分数增加迭代了多少次。
- 第 6 行 最终实际使用的质量值。
- 第 7 行 显示该样本在批次效应聚类后是否为异常值。
- 第 8 行 异常值的分数,显示有多少个覆盖数据点的 p 值小于 0.05。ps 它不能真正用于 QC 控制,因为较大的非整倍性会导致高比例的异常值。
The last line – fraction of outliers – shows how many coverage data points had p-value less than 0.05. It can not really be used for the QC control since having large aneuploidy will lead to high fraction of outliers.
最后一行(异常值的分数)显示有多少个覆盖数据点的 p 值小于 0.05。它不能真正用于 QC 控制,因为较大的非整倍性会导致高比例的异常值。
结果表格下:
| #chr | start | end | CN_change | loglikelihood | no_of_regions | length_KB | potential_AF | genes | qvalue |
|---|---|---|---|---|---|---|---|---|---|
| chr1 | 22315703 | 22336693 | 1 | 231 | 10 | 20.990 | 0.076 | - | 0.00000 |
| chr1 | 17248483 | 17275039 | 1 | 48 | 18 | 26.556 | 0.152 | - | 0.00419 |
| chr1 | 25599018 | 25655627 | 1 | 205 | 11 | 56.609 | 0.127 | - | 0.00000 |
| chr1 | 17060426 | 17086502 | 3 | 18 | 10 | 26.076 | 0.114 | - | 0.04763 |
| chr1 | 110230453 | 110236367 | 4 | 25 | 9 | 5.914 | 0.253 | - | 0.01483 |
- #chr: 染色体;
- start: CNV的起始;
- end:CNV的终止;
- CN_change:检测到的变体的拷贝数;
- loglikelihood:与基线(二倍体或男性性染色体的 1 个拷贝)相比,检测到的特定区域的拷贝数的可能性。,对数似然 > 10 意味着支持替代拷贝数的证据强度“非常强”,但是,我们建议您保持更高的值(40 或至少 20),因为基因组覆盖度受到影响诸如短插入缺失等众多事件导致比对问题、批次效应、测序深度、技术假象等,而且基因组相当长,因此看到大的对数似然变化并不罕见。
- no_of_regions: 显示变体中包含多少个数据点。较长的变体通常更可信(但并非总是如此,例如,对loglikelihood值较小的长变体可能是误报)。
- length_KB:CNV的长度(start和end间的距离)
- potential_AF:显示特定区域的覆盖率异常较低/较高的频率(如果变体分别为删除/重复)。
- genes:输入的.bed文件包含基因信息时, 会填充 genes 信息。
- qvalue: Z-test的p值,高p值,则对应区域可能是 CNV多态性 区域。
******_cnvs.seg
| ID | chr | start | end | value | |
|---|---|---|---|---|---|
| 20240827__24B07832897 | chr1 | 22315703 | 22336693 | 231.01 | 1 |
| 20240827__24B07832897 | chr1 | 17248483 | 17275039 | 47.98 | 1 |
| 20240827__24B07832897 | chr1 | 25599018 | 25655627 | 205.2 | 1 |
******_cov.seg
记录了bed上每个区域的异常值和折算的拷贝数信息。
| ID | chr | start | end | variance | value |
|---|---|---|---|---|---|
| 20240827__24B07832897 | chr1 | 65498 | 65638 | 0.0934 | 1.98 |
| 20240827__24B07832897 | chr1 | 69036 | 70008 | 0.0604 | 2.3 |
| 20240827__24B07832897 | chr1 | 367648 | 368607 | 0.0836 | 2.24 |
| 20240827__24B07832897 | chr1 | 564537 | 564657 | 0.1381 | 2.14 |
| 20240827__24B07832897 | chr1 | 621085 | 622044 | 0.0839 | 2.17 |
软件使用
针对遗传检测
基础分析参数
1 | Rscript /ifstj2/B2C_RD_H1/Personal/liubo/3.Software/ClinCNV-master/clinCNV.R \ |
可选参数
- –normalOfftarget & –bedOfftarget
添加脱靶覆盖率(不会大幅提高灵敏度,因为种系 CNV 很少与脱靶窗口一样长,但可以提高特异性,有效去除彼此远离的探针形成的 CNV,必须提取足够大的脱靶覆盖率样本数,但不一定是normal.cov文件中的所有样本)。
e.g.:--normalOfftarget normalOff.cov --bedOfftarget bedFileOff.bed - –scoreG
平衡灵敏度和特异性(增加阈值–scoreG 会导致更高的特异性和更低的灵敏度,反之亦然,默认值为 20)。
e.g.:--scoreG 60 - –lengthG
增加或减少检测到的变体的最小长度(默认 = 2 个数据点或更大,实际数据点数量实际上是您在此处指定的 +1,0 表示至少 1 个数据点)。
e.g.:--lengthG 0 - –maxNumGermCNVs & –maxNumIter
增加您期望从该样本中获得的 CNV 数量(默认 = 10000,但对于 WGS 样本,最好将其设置为 1000 甚至更大,当超过此阈值时,质量阈值会增加,并且样本会重新分析多次(由 –maxNumIter 指定)
e.g.:--maxNumGermCNVs 2000 --maxNumIter 5 - –minimumNumOfElemsInCluster 50
将大队列分成几个相似样本的集群,以进行更准确的参数估计并加快工具的速度。您指定集群的最小大小(我们建议将其保持在 20 到 100 之间):
e.g.:--minimumNumOfElemsInCluster 50 - –numberOfThreads 4
通过使用多个核心来加速分析过程中的某些部分,以提高分析速度。
e.g.:--numberOfThreads 4
Polymorphic CNVs
人群频率大于 2.5% 的 CNV 通常更难以使用常规方法检测。因此,我们使用高斯混合模型检测它们。这是同一脚本中的单独例程。要调用此类变体,您需要指定
- –polymorphicCalling YES
如果提供的参数不是 YES 而是带有多态区域坐标的.bed文件路径, ClinCNV将在调用过程中忽略他们。
嵌合体
嵌合体CNV的检测范围为 1.1 到 2.9,步长为 0.05。
- –mosaicism
软件原理介绍
因为目前预期用途是进行胚系的WES应用评估,所以相关方法说明参考的 2022年的ClinCNV: multi-sample germline CNV detection in NGS data
方法检测整体是基于深度的,算法可以比较容易的将等位基因频率纳入考量,但是文章认为等位基因频率对体系CNV的帮助比较大,但是对于胚系CNV检测作用并没有体系那么大。
method
检测方法整体分为两部分:
- 数据的标准化;
- CNV的Calling(这部分基于作者2019年发布的前一篇文章ClinCNV: novel method for allele-specific somatic copy-number alterations detection。
标准化
在这边文章所述方法中,作者针对标准化进行了调整,来实现以下目的(源于补充材料):
- 构建bin文件,分析的区段单元,通常是120bp,可以是WES富集捕获的区段,也可以是其他有依据的划分方式。(前期准备)
- 针对每个bin文件,统计reads的数目。(前期准备)
- 利用平方根转换$f(x)=\sqrt(x)$,根据(GC-content, region-length based, variance stabilization)进行方差稳定的数据标准化。
CNV检测
- 在所有样本中,寻找和目标样本的覆盖度相似度最高的样本,根据coverage 对样本进行聚类。
- 进行样本间的标准化,对聚类到一起的样本使用覆盖度的中位值进行标准化。
- 对统计模型进行参数估计用来描述不同的拷贝数状态(针对每个样本的每个区域)。
- 构建似然矩阵,矩阵的行对应预先定义的拷贝数状态,矩阵的列对应我们在 step1 中定义的bin的数目。
- 对bin进行片段化,并进行结果检出
- 使用各种QC矩阵进行注释,并对结果进行可视化。
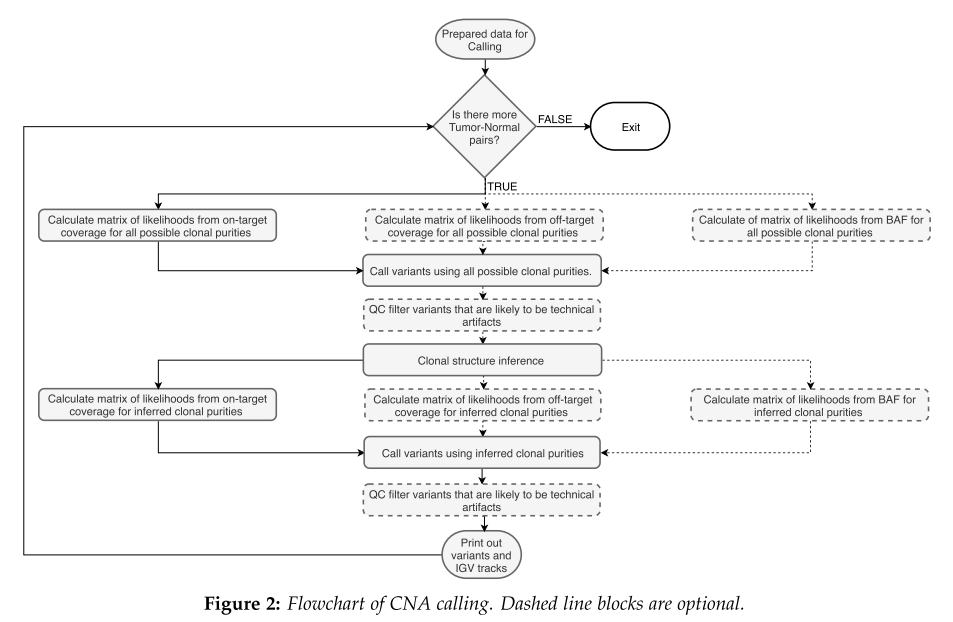
引自2019年发表的第一篇文章
1 | graph TB; |